Great Lakes Bioinformatics Conference 2012
Travel
Leading Professional Society for Computational Biology and Bioinformatics
Connecting, Training, Empowering, Worldwide

FASEB will provide two categories of MARC travel fellowships for the 2012 Great Lakes Bioinformatics Conference. Deadline April 3, 2012.
Please read the information at the FASEB website for eligibility.
FASEB Award Eligibility General Information Page: www.faseb.org/Marc/MARC-and-Professional-Development/Travel-Awards/Travel-Awards--Scientific-Meetings-and-Conferences.aspx
Faculty/Mentors and Students Travel Award: Each award provides funding for one faculty member/mentor and up to two students or post- baccalaureates from a minority institution to participate in one of the selected national meetings or conferences. The faculty/mentor must nominate up to two students and/or post-baccalaureates who are pursuing research careers in a life sciences discipline. The application packet must be submitted by the Faculty/Mentor listed on the application form. We do not accept Faculty/Mentor and Students Travel Award applications from students.
Poster/Platform (Oral) Presenter Travel Award: Each award provides funding for one underrepresented minority* graduate/ undergraduate student, post-baccalaureate or postdoctoral fellow who is 1st author and has been selected to give an oral or poster presentation at one of the selected national meetings or conferences.
[TOP]
All meetings are being held at the Michigan League Building, University of Michigan
http://uunions.umich.edu/league/maps/floorplans/
| go directly to: [Wednesday - May 16] [Thursday - May 17] | ||||
|
|
||||
| MONDAY - May, 14 2012 | ||||
| 4:00pm - 6:00pm | Registration (Concourse Room) | |||
| 4:00pm - 6:00pm |
Group Hike Nichols Arboretum www.lsa.umich.edu/mbg/see/nicholsarboretum.asp Nichols Arboretum "the Arb" is a 123 acre living museum set in the hills adjacent to the UM main campus. Originally designed by OC Simonds in 1906. Long views are framed by the Huron River Valley and unique plants. The Total hiking trail distance is just over 2 miles, with numerous points of interest and historical areas as well as viewing points of the surrounding areas. We will follow self-guided tours in small groups depending on the number of people who join us. We will meet at the GLBIO Registration Area in the Michigan League (2nd floor, Concourse), at 4 pm and travel as a group to the hike start. If you wish, you can meet us directly at the Nichols Arboretum, Washington Heights entrance (1610 Washington Heights; www.lsa.umich.edu/mbg/files/ArbGuide.pdf). We will be arriving there around 4:15pm - 4:20pm. |
|||
|
|
||||
| TUESDAY - May 15, 2012 | ||||
| Mendelssohn Theatre | Vandenberg Room | Hussey Room | ||
| 8:00am - 5:00pm | Registration | |||
| 9:00am - 11:00am |
Tutorial 1 High Performance Computing in Massive Omics Data Ma, Ping Univ. of Illinois |
Tutorial 2 RNA-Seq Approach to Study Gene Expression Wijeratne, Asela Ohio State Univ. |
Tutorial 3 Using New Tools for Predicting 3D Structural Motifs in Non coding RNAs Leontis, Neocles Bowling Green State Univ. |
|
| 11:00am - 11:10am | Break | |||
| 11:10am - 11:30am |
Industry Track 1 Bioteam (Mendelssohn Theatre) |
|||
| 11:30am - 12:45pm | Lunch Break (lunch on own) | |||
| 12:45pm - 2:45pm |
Tutorial 4 An Informatics Platform for Processing Cancer Jin, Victor Ohio State Univ. |
Tutorial 5 Accessing Free, Large-scale Computation and Data Resources for Bioinformatics through the eXtreme Science and Engineering Discovery Environment (XSEDE) Blood, Philip Carnegie Mellon Univ. |
Tutorial 6 Structural Variation Detection Using Next-generation Sequencing Hayes, Matthew Case Western Reserve Univ. |
|
| 2:45pm - 3:00pm | Break | |||
| 3:00pm - 5:00pm |
Tutorial 7 Practical Biological Network Visualization and Analysis Jurisica, Igor Univ. of Toronto |
Tutorial 8 Knowledge-based Interpretation of High throughput Data Farfan, Fernando Univ. of Michigan |
Tutorial 9 RNA 3D Structural Motifs: Definition, Analysis, and Prediction Laing, Christian Wilkes Univ. |
|
| 5:00pm - 5:20pm |
Industry Track 2 Cambridge Computer (Mendelssohn Theatre) |
|||
| 5:30pm - 7:30pm |
Poster Session A and Reception (Ballroom) Poster Session List by Presenter Last Name Poster Session List By Number |
|||
|
|
||||
| WEDNESDAY - May 16, 2012 | ||||
| Mendelssohn Theatre | Hussey Room | |||
| 8:00am - 5:00pm | Registration (Concourse Room) | |||
| 9:00am - 10:00am |
Keynote 1 Learning Population Histories from Genome Variation Data Russell Schwartz, Professor Department of Biological Sciences and Lane Center for Computational Biology Carnegie Mellon University Pittsburgh, PA - USA |
|||
| 10:00am - 10:10am | Break | |||
| 10:10am - 10:30am |
Industry Track 3 Compendia Bioscience |
|||
| 10:30am - 12:00pm |
WA1 Oral Presentations Gene Regulations and Transcriptiomics OP1, OP2, OP3, OP4, OP5, OP6 |
WA2 Oral Presentations Protein Structure & Function, Databases and Ontologies OP7, OP8, OP9, OP10, OP11, OP12 |
||
| 12:00pm - 1:30pm |
Lunch Break (lunch on own) |
|||
| 1:30pm - 2:30pm |
Keynote 2 Pathogen Diversity from an Ecological Perspective Mercedes Pascual, Professor Department of Ecology and Evolutionary Biology Affiliate of the Center for Computational Medicine and Bioinformatics University of Michigan Ann Arbor, MI - USA |
|||
| 2:30pm - 4:00pm |
WP1 Oral Presentations Evolution & Comp. Genomics, Population Genomics OP13, OP14, OP15, OP16, OP17, OP18 |
WP2 Oral Presentations Protein Interaction and Molecular Networks, Metagenomics OP19, OP20, OP21, OP22, OP23, OP24 |
||
| 4:00pm - 4:15pm | Break | |||
| 4:15pm - 5:15pm |
S1 - Special Session Proteins in Motion: Computational Studies in Protein Flexibility and Dynamics |
S2 - Special Session Computational Proteomics: Understanding Network Perturbations in Cancer |
||
| 5:15pm - 7:15pm |
Poster Session B and Reception Poster Session List by Last Name Poster Session List By Number |
|||
| 7:30pm - 10:00pm |
7:30pm - 8:30pm UMICH Museum Art Viewing ................................... 8:30pm to 10:00pm Dinner and Keynote 3 ................................... Designing Bioinformatics for the Wetware. Usability Challenges with Massive Amounts of Data. Howard Cash, President Gene Codes Corporation Gene Codes Forensics, Inc. Ann Arbor, MI - USA |
|||
|
|
||||
| THURSDAY - May 17, 2012 | ||||
| Mendelssohn Theatre | Hussey Room | |||
| 8:00am - 12:00pm | Registration (Concourse Room) | |||
| 9:00am - 10:00am |
S3 - Special Session Creating Teams for Bioinformatics |
Ohio Bioinformatics Consortium Meeting | ||
| 10:00am - 10:15am | Break | |||
| 10:15am - 12:00pm |
RA1 Oral Presentations - Sequence Analysis OP25, OP26, OP27, OP28, OP29, OP30, OP31 |
RA2 Oral Presentations Algorithm & Machine Learning, Bioimaging, Disease Models & Epidemiology OP32, OP33, OP34, OP35, OP36, OP37, OP38 |
||
| 12:00pm-1:00pm |
Keynote 4 Mutation, Drift, and the Evolution of Subcellular Features Michael Lynch, Professor Department of Biology Indiana University Bloomington, IN - USA |
|||
| 1:00pm - 1:15pm | Adjourn | |||
|
|
||||
| go directly to: [TOP] [Wednesday - May 16] [Thursday - May 17] | ||||
 |
Howard Cash, President Gene Codes Corporation Gene Codes Forensics, Inc. Ann Arbor, MI - USA Biosketch (.pdf) Title: Designing Bioinformatics for the Wetware. Usability Challenges with Massive Amounts of Data. Abstract: Gene Codes Corporation began producing DNA sequencing tools that dominated the commercial market starting in the mid-1990s. Starting in 1997 and especially after 9/11, 2001, the company produced tools that completely changed the standard for DNA analysis software in forensic crime labs around the world. In neither case did success depend on new discoveries or dramatically more sensitive pattern detection algorithms. Instead, the Sequencher and M-FISys programs, respectively, raised the game on usability for the bench scientist. For people in bioinformatics, the changes in data scale have been dramatic over the last several years. How do we make that information available to the people who will benefit from it? If a consumer could truly get a full genome in a matter of seconds as portrayed in the dystopian movie, GATTACA, surely they would not want a listing of all of the individual data points. So where might bioinformatics professional focus in the future? We will briefly discuss where bioinformatics has come in the days since MolGen, UWGCG and IntelliGenetics and some of the principles that can help tool developers who focus on the data present that information so that end users can grasp its content. [TOP] [Return to Full Agenda Page] |
 |
Michael Lynch, Professor Department of Biology Indiana University Bloomington, IN - USA CV: www.bio.indiana.edu/faculty/directory/profile.php?person=milynch Title: Mutation, Drift, and the Evolution of Subcellular Features Abstract: Understanding the mechanisms of evolution and the degree to which phylogenetic generalities exist requires information on the rate at which mutations arise and their effects at the molecular and phenotypic levels. Although procuring such data has been technically challenging, high-throughput genomic sequencing is rapidly expanding our knowledge in this area. Most notably, information on spontaneous mutations, now available in a wide variety of organisms, implies an inverse scaling of the mutation rate (per nucleotide site) with the effective population size of a lineage. The argument will be made that this pattern naturally arises as natural selection pushes the mutation rate down to a lower limit set by the power of random genetic drift rather than by intrinsic molecular limitations on repair mechanisms. Additional support for this idea derives from the relative levels of efficiency of DNA polymerases and mismatch-repair enzymes in eukaryotes relative to prokaryotes. This drift-barrier hypothesis has general implications for all aspects of evolution, including the performance of enzymes and the stability of proteins. The fundamental assumption is that as molecular adaptations become more and more refined, the room for subsequent improvement becomes diminishingly small. If this hypothesis is correct, the population-genetic environment imposes a fundamental constraint on the level of perfection that can be achieved by any molecular adaptation. It also implies that effective neutrality is the expected outcome of natural selection, an idea first suggested by Hartl et al. in 1985. Although generally viewed as an independent process, mutation also operates as a weak selective force, thereby playing a central role in “nearly neutral” hypotheses in evolution. Most notably, genes and proteins with more complex structures are subject to higher rates of mutational degeneration simply because they are larger mutational targets. However, because the mutation rate is very low at the nucleotide level, the efficiency of such mutation-associated selection becomes of diminishing significance in populations with small effective sizes. Thus, mutationally hazardous genomic and gene-structural features, which may or may not be adaptive, are expected to passively arise in lineages with small effective sizes. This general principle, the mutational-hazard theory, will be illustrated with examples including: 1) the differential expansion of intron numbers in various phylogenetic lineages; and 2) the diversification of protein-architectural features. [TOP] [Return to Full Agenda Page] |
 |
Mercedes Pascual, Professor Department of Ecology and Evolutionary Biology Affiliate of the Center for Computational Medicine and Bioinformatics University of Michigan Ann Arbor, MI - USA CV: www.lsa.umich.edu/eeb/directory/faculty/pascual Title: Pathogen Diversity from an Ecological Perspective Abstract: Sequence data is becoming increasingly available for many pathogens in time and space. This presents the opportunity to describe their population structure at different scales and to address the role of this structure for epidemiology. In particular, the genetic diversity associated with antigenic phenotypes, the variation recognized by the immune system, is of interest. Here, conceptual models with a basis in competitive ecological interactions provide a basis to understand whether immune selection (competitive interactions between strains mediated by cross-immunity) structures pathogen populations and how this structure influences in turn the transmission dynamics of pathogens. In this talk, I present two examples from our work on Plasmodium falciparum malaria and H3N2 influenza respectively, with individual-based models of transmission that track the history of infection of individual hosts and the genetic relatedness of the pathogen. Empirical analyses and considerations on testing resulting predictions on this ‘strain theory’ are discussed. [TOP] [Return to Full Agenda Page] |
 |
Russell Schwartz, Professor Department of Biological Sciences and Lane Center for Computational Biology Carnegie Mellon University Pittsburgh, PA - USA CV: www.cmu.edu/bio/faculty/schwartz.html Title: Learning Population Histories from Genome Variation Data Abstract: High throughput sequencing technologies have made it possible to assemble vast libraries of genetic variation data describing how genomes differ from one person to another. Such data implicitly encode a history of our species at a population level due to the gradual accumulation of mutations in our genomes as we have developed from an initial founder population to the many diverse subpopulations that comprise our species today. Better characterizing this history could be useful not only to basic research but also to important practical problems in improving the effectiveness of genotype/phenotype association studies and better understanding how we have adapted to historical disease threats or environmental changes. Reconstructing this population history from the indirect evidence of random mutations is a challenging computational problem, but also an interesting problem from a computational biologist’s perspective because of a long history of different approaches reflecting different biological perspectives and drawing on different classes of computational methods. In this talk, we will examine new strategies for characterizing population history that attempt to synthesize some of these different perspectives into a more complete model of our history as a species. This work seeks to combine discrete algorithms and operation research methods arising from the field of phylogenetics with machine learning and statistical sampling methods arising from statistical genetic models of population substructure. We will examine the motivation behind these approaches and explore their development from a computational perspective. We will then see what we can learn by applying such methods to real data. In the process, we will see some of the many ways diverse computing paradigms can contribute to current research in human genetics. [TOP] [Return to Full Agenda Page] |
General Information:
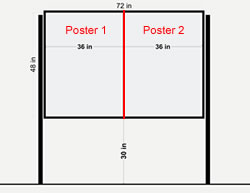
Schedule:
| DAY/DATE: | TIME: | ACTIVITY/LOCATION |
| Tuesday: May 15, 2012 | 2:00 pm to 4:00 pm | POSTER SESSION A PRESENTERS SET UP POSTERS (MAXIMUM poster dimensions are 46 inches high x 35 inches wide) |
| Tuesday: May 15, 2012 | 5:30pm to 7:30pm | POSTER SESSION A (authors present) See above for location Session A Presenters please remove your posters following the session so Session B presenters may set up |
| Wednesday: May 16, 2012 | 9:00 am to 4:00 pm | POSTER SESSION B PRESENTERS SET UP POSTERS (MAXIMUM poster dimensions are 46 inches high x 35 inches wide) |
| Wednesday: May 16, 2012 | 5:15pm to 7:15pm | POSTER SESSION B (authors present) See above for location (Authors please remove posters from boards at end of session) |
FURTHER QUESTIONS
GLBIO Conference Coordinator
Stephanie Hagstrom
This email address is being protected from spambots. You need JavaScript enabled to view it.
352-665-1763
[TOP]
Call for submissions of Poster Abstracts is now closed.
| KEY DATES | |
| March 15, 2012 | Call for Poster Abstracts |
| March 22, 2012 | Acceptance Notification for Poster Abstracts |
| May 4, 2012 | Registration Deadline for Poster Presenters |
Poster abstracts are solicited in all areas that involve the application of advanced computational methods to significant problems in biology or medicine.
Solicited Topic Areas:
• Algorithm Development and Machine Learning
• Bioimaging
• Databases and Ontologies
• Disease Models and Epidemiology
• Evolution and Comparative Genomics
• Gene Regulation and Transcriptomics
• Mass Spectrometry and Proteomics
• Metagenomics
• Population Genomics
• Protein Interactions and Molecular Networks
• Protein Structure and Function
• Sequence Analysis
• Text Mining
How to Submit:
Participants interested in presenting a poster at GLBIO are required to submit a 250-word abstract and scientific justification (why this work fits with the GLBIO community). Accepted abstracts will be printed on the conference website.
Conference Proceedings
Poster Presentations will NOT be considered for publication as part of the conference proceedings, only oral presentations.
Conference Registration
GLBIO conference registration for accepted oral and poster presenters must be completed by May 4, 2012 to remain eligible to present at GLBIO. Payment of the conference registration fee is the responsibility of the presenter.
>>CLICK HERE<< for the online submissions of poster abstracts.
Call for submissions of Oral Presentation Abstracts is now closed. Submissions for Poster Abstracts still available until March 15, please click here if you would like to submit a poster abstract. Please contact This email address is being protected from spambots. You need JavaScript enabled to view it.
The GLBIO 2012 Conference invites the submission of abstracts for 15-minute research oral presentation.
| KEY DATES | |
| Jan. 20, 2012 | Call for Oral Presentation Abstracts Closes |
| Mar 2, 2012 | Acceptance Notification for Oral Presentations |
| Apr 6, 2012 | Full Manuscripts (for proceedings) Due |
| May 15-17 | GLBIO Conference, Ann Arbor, MI |
Original oral presentation abstracts (including significant works-in-progress) are solicited in all areas that involve the application of advanced computational methods to significant problems in biology or medicine.
Solicited Topic Areas:
• Algorithm Development and Machine Learning
• Bioimaging
• Databases and Ontologies
• Disease Models and Epidemiology
• Evolution and Comparative Genomics
• Gene Regulation and Transcriptomics
• Mass Spectrometry and Proteomics
• Metagenomics
• Population Genomics
• Protein Interactions and Molecular Networks
• Protein Structure and Function
• Sequence Analysis
• Text Mining
How to Submit:
Participants interested in presenting at GLBIO are required to submit: (a) 250-word scientific justification (why this work fits with the GLBIO community), (b) 250-word abstract (this appears in the conference agenda), and (c) 1-page PDF summary of research to be presented (this will aid in the selection of high-quality oral presentations). Accepted oral presentation abstracts will be printed in the conference program and included on the conference website.
Conference Proceedings![]() BMC Proceedings will publish the proceedings of the Great Lakes Bioinformatics Conference. Authors may elect to submit a (7-10 page) manuscript for
consideration by April 6, 2012 (CLICK HERE for submission page).
BMC Proceedings will publish the proceedings of the Great Lakes Bioinformatics Conference. Authors may elect to submit a (7-10 page) manuscript for
consideration by April 6, 2012 (CLICK HERE for submission page).
The fee for publication is £680 (British
Pound Sterling) and will be invoiced directly to each author by the
publisher.
Following a full review, accepted manuscripts will move through the following publication timeline:
| Author Notification | 03/02/2012 |
| Submission of Manuscripts for Proceedings |
04/06/2012 |
| Author Notification of Review Decision | 05/21/2012 |
| Final Manuscript Submitted | 06/15/2012 |
Manuscripts submitted for publication in the proceedings must be original (not already published elsewhere).
Please note the acceptance of the oral presentation does not guarantee publication of the manuscript; all submitted manuscripts will undergo a full peer review.
Manuscripts shall be prepared according to the "Instructions for preparation of manuscripts for publication in supplements to BioMed Central journals".
Instructions:
Conference Registration
GLBIO conference registration for accepted oral and poster presenters must be completed by May 4, 2012 to remain eligible to present at GLBIO. Payment of the conference registration fee is the responsibility of the presenter.
![]()
