In-person Tutorials (All times BST)
Virtual Tutorials: (All times BST) Presented through the conference virtual platform
Tutorial IP1: Machine Learning for Omics: Best practices and Real-Life Insights with TidyModels SOLD OUT
Room: 11A
Date: July 20, 2025
Start Time: 09:00
End Time: 18:00
Organizer:
Jamie Soul
Speakers:
Jamie Soul, University of Liverpool
Eva Caamano Gutierrez, University of Liverpool
Anthony Evans, University of Liverpool
Max Participants: 30
Description
Omics data analysis presents unique challenges due to its high dimensionality and complexity. Supervised machine learning (ML) offers powerful tools for gaining insights from these data but currently faces a crisis of reproducibility due to poor adherence to best practices when undertaking feature selection, model evaluation, and needs for further interpretability.
This full-day tutorial introduces participants to the common pitfalls and best practices of applying ML to omics research. It exemplifies good practice through example using the Tidymodels framework for ML workflows in R, tailored to omics applications. The course will feature a mixture of lectures, quizzes, real-life coding tutorials and hands-on practicals with 1-1 support. Example applications will illustrate regression analysis with methylation clocks, gene prioritisation and classification with cancer biomarker discovery.
Special attention will be paid to challenges in working with highly multivariate data and integrating various data types as well as providing tips to extract meaningful insights from complex data. Beginner-level R skills are required, and attendees will leave with practical skills to apply Tidymodels to their own datasets.
Learning Objectives
- Understand the challenges and pitfalls of using supervised machine learning on omics data, including reproducibility, overfitting, and feature selection.
- Ability to critically appraise published examples as well as gain familiarity with reporting practices such as DOME.
- Gain hands-on experience using Tidymodels to implement machine learning workflows in R.
- Learn techniques for feature selection, dimensionality reduction, and inclusion of network-based information.
- Explore interpretable machine learning approaches to improve biological insights.
- Develop skills to apply Tidymodels for methylation clock modeling, gene prioritisation and mechanistic biomarker discovery.
Intended Audience and Level
This course is designed for researchers with at least a basic understanding of R programming and an interest in applying machine learning to omics data analysis. Prior exposure to omics data is beneficial but not mandatory. The course will provide a gentle introduction to relevant concepts while focusing on practical skills development. No prior experience with Tidymodels or advanced machine learning techniques is required. The tutorial is tailored to beginners and those looking to expand their skills in machine learning data analysis using R.
Schedule
| 09:00-09:45 |
Lecture: Introduction to Machine Learning in Omics
- Introductions and overview of the course structure and learning objectives
- Introductory lecture on ML principles, applications and common pitfalls in application to omics data, including “crisis of reproducibility”, high dimensionality, and interpretability challenges
- Introduction to reporting recommendations (e.g. DOME, FAIR adherence) and quiz to reinforce key concepts
Eva Caamano Gutierrez |
| 09:45-10:00 |
Hand-on: Critical appraisal of published examples
Identifying good practice and areas of development. Expand familiarity with best reporting practices. |
| 10:00-10:45 |
Tidymodels Framework: A Practical Introduction
A follow along tutorial covering:
- Introduction to Tidymodels: Overview of the Tidymodels ecosystem and its core packages and how tidymodels streamlines the end-to-end machine learning workflow
- Understanding and Avoiding Data Leakage: Explanation of data leakage and why it can compromise model validity and examples of best practices
- Specifying Preprocessing Recipes: How to define reusable preprocessing pipelines using the recipes package
- Model Fitting with Parsnip: Introduction to different model types and how Tidymodels supports multiple ML algorithms
- Model Evaluation with Yardstick: Best practices for evaluating model performance using appropriate metrics and hands on demonstration
Dr Jamie Soul |
| 10:45-11:00 |
Coffee Break |
| 11:00-12:30 |
Hands-on: Explore Tidymodels basics with a provided Quarto notebook
Participants will work through a quarto notebook at their own pace implementing a simple classification model using the tidymodels framework based on the previous demonstration. |
| 12:30-13:00 |
Lecture: Strategies for Managing Large Omics Datasets
This lecture will provide strategies for handling the challenges posed by large omics datasets. Topics will include class imbalance, techniques for reducing dimensionality, and feature selection methods. Participants will learn how these methods can enhance model performance and reveal key biological insights. The session will conclude with a quiz to test understanding and reinforce key concepts discussed during the lecture.
Jamie Soul |
| 13:00-14:00 |
Lunch Break |
| 14:00-15:15 |
Hands-on: Using Tidymodels to build a methylation clock
In this interactive session, participants will learn how to use the Tidymodels framework to build a cross-validated regression model. The focus will be on predicting the age of an organism using methylation array data, providing a practical introduction to model building, validation, and analysis in the context of biological data. |
| 15:15-15:30 |
Lecture: A short snapshot of networks and pathways
Introduction to networks and considerations for inclusion for ML
Anthony Evans |
| 15:30-16:00 |
Hands on: Enhancing Machine Learning with Biological Context: Integrating Networks and Pathways
This practical session will explore strategies for incorporating additional biological information, such as networks and pathway data, into machine learning models. Participants will examine how these methods can enhance model performance and biological relevance when applied to the task of gene prioritsation. |
| 16:00-16:15 |
Coffee Break |
| 16:15-16:30 |
Lecture: a short introduction to gaining model interpretability
Introduction to interpreting models and tradeoffs between performance and interpretability.
Anthony Evans |
| 16:30-17:30 |
Hands On: Identifying biological mechanisms with ML
With examples applied to cancer biomarker discovery, participants will explore how interpretable machine learning (ML) techniques can be used to gain insights into biological mechanisms. The session will provide a practical introduction to key interpretability tools, such as SHAP (SHapley Additive exPlanations) values which help understand the relationship between input features and model predictions. |
| 17:30-18:00 |
Wrap-Up and Discussion Review of key concepts, Q&A, and discussion of applications to their own research. |
- top -
Tutorial IP2: Massively parallel reporter assays in functional regulatory genomics and as part of the IGVF data resource
Room: 03A
Date: July 20, 2025
Start Time: 09:00
End Time: 18:00
Organizer:
Martin Kircher
Speakers:
Martin Kircher, University of Lübeck and Kiel University
Max Schubach, Berlin Institute of Health at Charité
Arjun Devadas, University of Lübeck and University Hospital Schleswig-Holstein
Kilian Salomon, Berlin Institute of Health at Charité
Max Participants: 100
Description
This tutorial is designed to empower bioinformatics researchers with the knowledge and skills to effectively utilize Massively Parallel Reporter Assays (MPRAs) data in their work. MPRAs are gaining wider applications across the functional genomics community and are used as part of the Impact of Genomic Variation on Function (IGVF) Consortium. IGVF is a collaborative research initiative funded by the NHGRI that aims to systematically study how genomic variations affect genome function and, consequently, phenotypes. By integrating experimental and computational approaches, IGVF seeks to map and predict the functional impacts of genetic variants, providing a comprehensive catalog of these effects.
This tutorial provides a thorough introduction in MPRAs and IGVF data resources, practical training on MPRA data, and insights into advanced analysis methods for such data. Participants will gain an understanding of MPRA experiments, including their various experimental designs and the rationale for using them in functional genomics. This will involve learning the process of associating tags/barcodes with sequences incorporated in the reporter constructs from raw sequencing reads and counting barcodes from DNA sequencing and RNA expression. The tutorial will guide participants through data processing using MPRAsnakeflow, a streamlined snakemake workflow developed with IGVF for efficient MPRA data handling and QC reporting. Statistical analysis for sequence-level and variant-level effect testing of MPRA count data will be introduced using BCalm, a barcode-level MPRA analysis package developed as part of our IGVF efforts.
Further, the tutorial will provide a starting point for training (deep learning) sequence models on MPRA data and related functional genomics datasets. Participants will learn how to extract meaningful insights from their datasets by investigating the sequence activity relationship and extracting important sequence motifs. By integrating these topics and methods, participants will leave the tutorial equipped with both theoretical knowledge and practical skills necessary for analyzing and using MPRA data effectively.
Learning Objectives
- Introduction to IGVF and how to access its data.
- MPRA Experiments: Explanation of experimental design, including the association of barcode/tag sequences to tested regulatory sequences and variants.
- Step-by-step data processing using MPRAsnakeflow, including construction of count tables and interpretation of quality control metrics.
- Statistical analysis of DNA and RNA barcode counts using BCalm.
- Applications of MPRA data like identifying active regulatory regions and significant variant effects, optimising cell-type-specific expression through synthetic sequences.
- Sequence-based modeling of regulatory sequence activity, i.e. using deep learning models to predict open chromatin and MPRA activity, investigate transcription factor motif importance and composition.
Intended Audience and Level
Beginner
Schedule
| 09:00-09:15 |
Welcome and introduction to the schedule |
| 09:15-09:30 |
Tutorial materials access and setup of the Google CoLab environment
Participants that fail with setting up their access und CoLab will be brought outside of the seminar room and helped by other speakers while the theoretical introductions commence |
| 09:30-09:50 |
Lecture: IGVF and its data access
Introduction to the IGVF mission and how to access its data |
| 09:50-10:15 |
Lecture: Fundamentals of MPRA Experiments
Explanation of experimental design, including the association of barcode/tag sequences to tested regulatory sequences and variants. |
| 10:15-10:45 |
Coffee break |
| 10:45-11:05 |
Hands-on: Association of Barcode/Tag Sequences
Participants will learn how to associate barcode/tag sequences with designed oligos using the association step of the MPRAsnakeflow pipeline on a subsetted/small dataset. |
| 11:05-11:30 |
Hands-on: Count Sequencing Analysis
We will demonstrate how to count barcodes in both DNA and RNA sequencing on a subsetted/small dataset. |
| 11:30-11:50 |
Discussion of QC metrics
We will discuss the QC parameters and plots returned by MPRAsnakeflow for the full data set, emphasizing the importance of accurate quantification in MPRA experiments. |
| 11:50-12:30 |
Hands-on: Data Analysis Steps (Regions and Variants)
We will walk through potential analytical steps for utilizing count tables and perform statistical analysis using BCalm to identify regions with high activity compared to other sequences in the assay. Additionally, we will highlight methods for identifying variants with significant differences between reference and alternative sequences within designed oligos. |
| 12:30-13:30 |
Lunch break |
| 13:30-13:50 |
Lecture: Modeling of regulatory activity with sequence models
We will provide a brief introduction and review of modeling efforts for sequence activity using gapped kmers, convolutional neural networks and language model approaches. |
| 13:50-14:30 |
Hands-on: Training a sequence based model
We will walk through the steps of training a model based on the DeepSTARR CNN architecture. |
| 14:30-14:50 |
Discussion of pre-training and fine-tuning approaches
We discuss the opportunity of pre-training convolutional layers on open chromatin data from multiple cell-types and fine-tuning these convolutions on MPRA data vs. using a language model like NT to train MPRA activity models. |
| 14:50-15:30 |
Hands-on: Interpreting models with in-silico mutagenesis
Supported by tools like TF-MoDISco for motif discovery, participants will investigate important transcription factor binding motifs using models trained on sequence data that predict activity. We will assess whether these motifs exert activating or repressing effects by comparing the activity of sequences with and without these identified motifs in the cell-type of interest. |
| 15:30-16:00 |
Coffee break |
| 16:00-16:40 |
Hands-on: Linking motifs to variant effects
We will identify transcription factor binding sites (TFBS) overlapping significant variants using motifs derived from model interpretation or available position weight matrices (PWMs) to support the interpretation of variant effects. |
| 16:40-16:50 |
Discussion of data limitations
We discuss the limitations of variant effects in various sequence contexts. We consider the effects of studying only few or related cell-types and the sensitivity of the available modeling approaches for cell-type effects. |
| 16:50-17:20 |
Participant questions and feedback |
| 17:20-17:30 |
Concluding remarks by the speakers |
- top -
Tutorial IP3: Genomic Variant Interpretation & prioritisation for clinical research
Room: 04AB
Date: July 20, 2025
Start Time: 09:00
End Time: 18:00
Organizer:
Aleena Mushtaq
Speakers:
Mallory Freeberg, EMBL's European Bioinformatics Institute (EMBL-EBI)
Aleena Mushtaq, EMBL's European Bioinformatics Institute (EMBL-EBI)
Diana Lemos, EMBL's European Bioinformatics Institute (EMBL-EBI)
James Stephenson, EMBL's European Bioinformatics Institute (EMBL-EBI)
Sarah Hunt, EMBL's European Bioinformatics Institute (EMBL-EBI)
Irene Lopez Santiago, Open Targets Bioinformatician (EMBL - EBI)
Genevieve Evans, EMBL's European Bioinformatics Institute (EMBL-EBI)
Max Participants: 40
Description
The interpretation of genetic variation is important for understanding human health and disease. Increased knowledge leads to societal benefits including faster disease diagnosis, a better understanding of disease progression, more efficient identification and prioritisation of drug targets for testing, resulting in overall better health outcomes for a population. Whilst the speed and cost of sequencing has reduced, the complexity of variant interpretation remains a bottleneck for understanding. This tutorial will explore the variety of annotations and techniques available to assess human variation and the implications of variant effects on human health and disease.
Learning Objectives
By the end of the tutorial, we expect participants will be able to:
- Explore human variation types in commonly used bioinformatics file formats
- Compare experimental methods for variant effect analysis and understand the specific strengths of each.
- Use bioinformatics knowledge bases to explore information about genetic variants and to contrast different approaches to see how they can be combined for thorough interpretation.
- Evaluate evidence from multiple sources supporting variant effect and impact in the context of research and study design
- Explore the impact of variant interpretation on clinical diagnostics and drug tractability.
Intended Audience and Level
Attendees should have an undergraduate degree / diploma level understanding of molecular biology, genetics, or biochemistry. It is a tutorial offered at the beginner level and no prior background in variant interpretation is required. Participants must be comfortable using web browsers. Laptop computers with an Internet connection will be required for the tutorial sessions.
Schedule
| 09:15-09:30 |
Lecture 1: Introduction - The challenge of variant interpretation
Variation in context of human health |
| 09:30-10:00 |
Lecture 2: - Genomic Annotation for variation datasets
Public annotation datasets, Variation sources, Transcript based annotations, Non-coding variation, Structural variation |
| 10:00-10:45 |
Hands-on 1: Annotating and predicting molecular variant effect
Variant prioritisation and scoring methods |
| 10:45-11:00 |
Break |
| 11:00-11:15 |
Lecture 3: Understanding Variant Effects Using Protein Structure
Protein position, interaction and complexes for variant interpretation, Alphafold3 and predicted structures |
| 11:15-11:45 |
Lecture 4: Understanding Variant Effects Using Protein Function
Combining functional, structural and population annotations to contextualise variant effects in proteins |
| 11:45-12:15 |
Hands-on 2: Using protein databases to investigate variant impact
Using structural information to interpret variant effect |
| 12:15-12:45 |
Lecture 5: Deep Mutational Scanning
Genome Editing for Variant Analysis |
| 12:45-14:00 |
Lunch Break |
| 14:00-14:30 |
Lecture 6: Utilising clinical data in variant prioritisation and classification, Applications fordisease research and genomic diagnosis, DECIPHER, G2P, and GWAS Catalog |
| 14:30-15:00 |
Lecture 7: Target tractability and drug associations
Target prioritisation for drug discovery, Case studies |
| 15:00-16:00 |
Group Projects
Hands on activity using bioinformatics resources for variant interpretation |
| 16:00-16:15 |
Break |
| 16:15-17:00 |
Group Projects
Hands on activity using bioinformatics resources for variant interpretation |
| 17:00-17:45 |
Presentations from groups
Present to peers to discuss ideas and future work |
| 17:45-18:00 |
Closing Remarks |
- top -
Tutorial IP4: Quantum Machine Learning for multi-omics analysis
Room: 03B
Date: July 20, 2025
Start Time: 09:00
End Time: 18:00
Organizer:
Aritra Bose
Speakers:
Aritra Bose, IBM Research
Hakan Doga, IBM Research
Akhil Mohan, Cleveland Clinic
Laxmi Parida, ISCB Fellow
Bryan Raubenolt, Cleveland Clinic
Filippo Utro, IBM Research
Max Participants: 50
Description
Single-cell and population-level multi-omics analyses have greatly enhanced our understanding of biological complexity. By integrating various types of biological data—such as genomics, proteomics, and transcriptomics, collectively known as multi-omics—these approaches have provided deep insights into the molecular mechanisms underlying complex diseases, both at the cellular level and across patient populations. As the size and complexity of multi-omics data continues to grow, the need to leverage emerging technologies such as artificial intelligence (AI) and quantum computing (QC) also grows. Recently, advances in QC have shown promise in solving real-world problems in machine learning and optimization in biomedicine, drug discovery, biomarker discovery, clinical trials, among other healthcare and life sciences objectives [1,2,3,4,5].
In this tutorial, participants will learn the fundamental concepts of QC, engage in hands-on experiments that apply classical machine learning (ML) techniques. They will also learn best practices for pre-processing multi-omics data in preparation for quantum machine learning (QML) tasks. Through a systematic evaluation of various data complexity measures and their impact on the performance of different ML and QML models, participants will gain insights into when to effectively utilize QML models. Additionally, they will explore quantum-classical hybrid workflows for ML, with a focus in biomedical data analysis.
Learning Objectives
- Understanding the fundamentals of quantum computing, including learning how to
implement algorithms in a quantum computer with quantum gates and circuits using
Qiskit.
- Practical experience of pre-processing multi-omics data and preparing it for a quantum
hardware experiment.
- Analyze machine learning methods on multi-omics data, understand their shortcomings
and review the impact of data complexity measures on ML models.
- How to apply QML models on multi-omics data.
- Learn design of experiments for biomedical data using quantum computers by gaining
an in-depth knowledge of quantum-classical hybrid workflows.
- Understand when to apply QML models and benchmark it with classical ML models.
Intended Audience and Level
This tutorial is aimed at computational biologists, bioinformaticians, clinicians, practitioners, data analysts, including early-career to senior researchers in the fields of healthcare and life sciences enthusiastic to learn about new frontiers of computational biology. There are very few prerequisites for the tutorial, listed as follows:
Schedule
| 09:00-09:20 |
Welcome Remarks and Introduction
|
| 09:20-10:00 |
Hands-on: Quantum computing fundamentals with Qiskit |
| 10:00-10:45 |
Current state of Quantum Machin Learning |
| 10:45-11:00 |
Coffee Break |
| 11:00-11:30 |
Data and Complexity measures |
| 11:30-12:00 |
Quantum Kernel methods |
| 12:00-13:00 |
Hands-on: Applying a Quantum-Classical machine learning benchmarking tool on omics data |
| 13:00-14:00 |
Lunch |
| 14:00-15:00 |
Hands-on: Implementing Quantum Kernel methods |
| 15:00-16:00 |
Hands-on: Execute the benchmarking tool on omics data |
| 16:00-16:15 |
Coffee Break |
| 16:15-17:15 |
Hands-on: Review results from the benchmarking tool |
| 17:15-17:45 |
Result Read-outs |
| 17:45-18:00 |
Furute Directions & Concluding Remarks |
- top -
Tutorial IP5: Introduction to Causal Analysis using Mendelian Randomisation
Room: 12
Date: July 20, 2025
Start Time: 09:00
End Time: 13:00
Organizer:
Handan Melike Dönertaş
Speakers:
Handan Melike Dönertaş, Fritz Lipmann Institute on Aging
Mark Olenik, Fritz Lipmann Institute on Aging
Tayyaba Alvi, Fritz Lipmann Institute on Aging
Max Participants: 20
Description
Mendelian randomisation (MR) is a method that uses genetic variation associated with an exposure (e.g., behaviours, biomarkers) to infer its causal effect on an outcome (e.g. health status). In statistical terms, it functions as an "instrumental variable" approach.
By mimicking the design of a randomised controlled trial through genetic inheritance, MR provides a framework for addressing confounding and reverse causation, making it a valuable tool in epidemiological and biomedical research.
This workshop offers a beginner-friendly introduction to the key concepts and assumptions underlying MR, such as the use of genome-wide association study (GWAS) data and the three key assumptions for valid instrumental variables: relevance, independence, and exclusion restriction. Participants will explore common challenges in MR analysis, including pleiotropy, population stratification, and measurement error while learning strategies to overcome these using advanced methods. The workshop also includes a two-hour hands-on session in which attendees will work with real-world data to conduct MR analyses using R. By the end of the session, participants will have a clear understanding of MR principles, the ability to critically evaluate MR studies, and practical skills to apply MR methods in their own research.
Learning Objectives
- Understand the key principles and assumptions underlying Mendelian Randomization.
- Identify potential challenges in MR studies, including biases and violations of assumptions.
- Familiarize with various MR methods and their applications (e.g., two-sample MR, IVW, MREgger, weighted median).
- Learn how to access and prepare genetic datasets for MR analysis.
- Conduct MR analyses using R, visualize and interpret results.
Intended Audience and Level
This workshop is designed for researchers and students new to MR or looking to incorporate it into their studies. No prior experience is required, although familiarity with R is required.
Schedule
| 09:00-09:45 |
Introduction to Mendelian Randomization
- Overview of MR and its applications
- Key assumptions: relevance, independence, and exclusion restriction
- Types of MR studies (one-sample, two-sample, bi-directional)
|
| 09:45-10:45 |
Challenges in Mendelian Randomization
- Horizontal pleiotropy and methods to address it
- Sample overlap and measurement error
- Population stratification and genetic heterogeneity
|
| 10:45-11:00 |
Coffee Break |
| 11:00-13:00 |
Hands-On Tutorial in R
- Preparing exposure and outcome GWAS data
- Conducting basic MR analysis (TwoSampleMR R package)
- Robust methods: MR-Egger, weighted median, and leave-one-out analysis
- Interpreting and visualizing results
|
- top -
Tutorial IP6: Hello Nextflow: Getting started with workflows for bioinformatics
Room: 11BC
Date: July 20, 2025
Start Time: 09:00
End Time: 13:00
Organizer:
Geraldine Van der Auwera
Speakers:
Geraldine Van der Auwera
Max Participants: 30
Description
Nextflow is a powerful and flexible open-source workflow management system that simplifies the development, execution, and scalability of data-driven computational pipelines. It is widely used in bioinformatics and other scientific fields to automate complex analyses, making it easier to manage and reproduce large-scale data analysis workflows.
This training workshop is intended as a “getting started” course for students and early-career researchers who are completely new to Nextflow. It aims to equip participants with foundational knowledge and skills in three key areas: (1) understanding the logic of how data analysis workflows are constructed, (2) Nextflow language proficiency and (3) command-line interface (CLI) execution.
Participants will be guided through hands-on, goal-oriented exercises that will allow them to practice the following skills:
- Use core components of the Nextflow language to construct simple multi-step workflows effectively.
- Launch Nextflow workflows locally, navigate output directories to access results, interpret log outputs for insights into workflow execution, and troubleshoot basic issues that may arise during workflow execution.
By the end of the workshop, participants will be well-prepared for tackling the next steps in their journey to develop and apply reproducible workflows for their scientific computing needs. Additional study-at-home materials will be provided for them to continue learning and developing their skills further.
Learning Objectives
We have delivered versions of this tutorial session at both online and in-person events previously (total of ~300 participants since May 2024), and have typically had to turn away would-be participants due to higher demand than we could accommodate. Post-event satisfaction surveys have typically shown the material was very well received. We are continuing to further expand and refine the material based on participant feedback.
Intended Audience and Level
Schedule
| 09:00-10:00 |
Hello World
Basic components and principles involved in assembling and running a Nextflow workflow.
- Learn the basics of the Nextflow syntax, how a workflow script is structured and how it can be modified
- Run a workflow for the first time, parse messages and logs produced during a run, and find outputs
- Use variables and key command-line parameters to control inputs, outputs and execution behavior
- Chain multiple steps together and handling transfer of data between steps
- Handle input and output files
|
| 10:00-10:20 |
Hello Containers
Using containers as a mechanism for managing software dependencies in the context of reproducible bioinformatics workflows. |
| 10:20-10:50 |
Hello Config
Setting up and managing a pipeline’s configuration to customize its behavior, adapt it to different environments, and optimize resource usage. |
| 10:50-11:05 |
Hello Modules
Using code modules to make pipeline development and maintenance more efficient and sustainable. |
| 11:05-11:20 |
Next steps
Overview of educational resources that participants can use to continue developing their Nextflow skills. Includes several domain-specific tutorials (currently Genomics, RNAseq in development) that provide a practical application of the concepts learned in this workshop to relevant use cases, and an orientation to the nf-core ecosystem of tools and pipelines that can be used out-of-the-box or as building blocks for customized solutions. |
- top -
Tutorial IP7: AI large cellular models and in-silico perturbation SOLD OUT
Room: 11BC
Date: July 20, 2025
Start Time: 14:00
End Time: 18:00
Organizer:
Xuegong Zhang
Speakers:
Xuegong Zhang, Tsinghua University
Chen Li, Tsinghua University
Erpai Luo, Tsinghua University
Mo Chen, Tsinghua University
Max Participants: 50
Description
Transformer-based large language models (LLMs) are changing the world. The capabilities they illustrated in sophisticated natural language, vision and multi-modal tasks have inspired the development of large cellular models (LCMs) for single-cell transcriptomic data, such as scBERT, Geneformer, scGPT, scFoundation, GeneCompass, scMulan, etc. After pretraining on massive amount of single-cell RNA-seq data agnostic to any downstream task, these transformer-based models have demonstrated exceptional performance in various tasks such as cell type annotation, data integration, gene network inference, and the prediction of drug sensitivity or perturbation responses. Such advancements, albeit still in their early stage, suggested promising revolutionary approaches for leveraging AI to understand the complex system of cells from extensive datasets beyond human analytical capacity. Especially, such models have made it possible to conduct in-silico perturbation on cells of various types to predict their responses to gene perturbations without doing experiments on the cells. These models provided prototypes of digital virtual cells that can be used to reconstruct and simulate live cells, which will revolutionize many aspects of future biomedical studies.
Although the community is high enthusiastic to these exciting progresses, the structures and algorithms of LCMs and other similar-scale AI models are mysterious to many people who were not equipped with relevant backgrounds. This tutorial will try to fill this gap. In the tutorial, we will begin from an introduction of basic principles of deep neural networks, and explain the basic structure and algorithm of the original Transformer for natural language tasks. We’ll show to the attendees how to build such models based on current machine learning platforms. Then we’ll introduce several successful ways to build large cellular models based on the basic Transformer model, and overview how such models are pretrained on single-cell RNA-seq data. We’ll show and let the attendees to practice how to use LCMs for basic tasks such as cell type annotation, and look into the specific application of LCMs on in-silico perturbation tasks. Attendees will engage in hands-on activities such as building basic transformer models and executing downstream single-cell tasks, including cell type annotation and in-silico perturbation. These activities will remove the mystery of LCMs for the attendees and help them better understand and feel how LCMs can be built and applied.
Learning Objectives
Attendees will build up their basic understanding of deep neural networks, Transformers, LLMs and LCMs. They will gain hands-on experiences on building such models and using existing models for downstream tasks. The particular knowledge and skill they’ll learn from this tutorial include:
- Understanding the basic principles of transformer models and large language models
- Knowing of the basic structure and components of transformer-based LCMs
- Experiencing building their own Transformer models
- Using pretrained LCMs cellular analyses on tasks such as cell label transfer and in-silico perturbation
- Forming a more complete picture of the current and future development of the field.
Intended Audience and Level
This tutorial is designed to be friendly for everyone who is interested in current AI but does not know much about how it works, including computational biologists, biologists, clinicians, or other professionals. For those who know something about AI and who had some pervious experiences on it, the tutorial will provide deeper understandings and hands-on experiences on advanced virtual cell applications of LCMs. Attendees are expected to have some basic knowledge of Python programming for completing the hands-on activities.
Schedule
| 14:00-14:30 |
Introduction to artificial neural networks and deep learning |
| 14:35-15:10 |
Introduction to language models and Transformers |
| 15:10-15:25 |
Coffee break |
| 15:25-15:55 |
Basic structure and components of transformer-based LCMs |
| 15:55-16:15 |
Hands-on: Build transformer model with Python
- Building a toy transformer model with PyTorch
- Using pretrained LCMs for cell type annotation
|
| 16:15-16:45 |
LLMs for in-silico perturbation |
| 16:45-17:00 |
Coffee break |
| 17:00-17:20 |
Hands-on: in-silico gene perturbation
- Model construction and data preparation
- In-silico gene perturbation with LCMs
- Evaluating the results
|
| 17:20-17:40 |
Discussions |
| 17:40-18:00 |
Summary |
- top -
Tutorial IP8: Representation Learning and Feature Engineering for Genomic Sequences Analysis SOLD OUT
Room: 12
Date: July 20, 2025
Start Time: 14:00
End Time: 18:00
Organizer:
Fabiana Goes
Speakers:
Fabiana Goes, The Rosalind Franklin Institute
Aparajita Karmakar, The University of Edinburgh and The Rosalind Franklin Institute, UK
Max Participants: 15
Description
Machine learning (ML) has been successfully applied in different omics problems, such as sequence classification in the field of genomics. The effectiveness of ML methods relies greatly on the selection of the data representation, or features, that extract meaningful information from sequences. Genomic sequences can be viewed as one-dimensional strings of successive letters representing nucleotides. However, to make these sequences compatible with ML methods, they must first be transformed into structured numerical representations, such as vectors or matrices. Traditional methods for sequence classification often rely on manually crafted or pre-defined features, which require domain expertise and may not fully capture the complexity of the underlying biological information. Recently, representation learning has emerged as a powerful alternative, enabling the automatic extraction of latent patterns directly from raw data and reducing the dependence on manually crafted features. In genomics, representation learning methods have been introduced to characterize DNA and RNA sequences. In genomics, techniques like Word2Vec, Convolutional Neural Networks (CNNs) and Large Language Models (LLMs) have demonstrated the ability to learn optimal sequence representations that effectively capture both local and global patterns in DNA and RNA sequences.
This tutorial provides a comprehensive introduction to feature engineering and representation learning for genomic sequences (DNA/RNA). Participants will explore traditional techniques for extracting features from genomic sequences, building a foundation in classical approaches. Furthermore, the tutorial will cover representation learning, introducing concepts such as embeddings and their applications. Topics include methods such as Word2vec and LLMs to obtain meaningful representations from genomic sequences. Through hands-on exercises and comparative analyses, attendees will learn to combine traditional feature engineering with representation learning approaches, developing practical skills and insights that are adaptable to diverse genomic research challenges. The goal is to offer participants the knowledge and tools to enhance genomic sequence analysis using different techniques for sequence representation.
Learning Objectives
- Participants will learn to analyze and extract numerical representations from genomic sequences (DNA/RNA) using both traditional and advanced techniques. Following the tutorial, they will be able to:
- Obtain an overview of the advancement of methods, from traditional feature extraction methods to representation learning techniques, and understand their impact on genomic data analysis.
- Understand the differences between genomic sequence embeddings and handcrafted features, exploring their roles in capturing complex patterns and insights within genomic sequences.
- Learn how to extract and engineer features from genomic sequences, focusing on key aspects like k-mers, sequence composition, and numerical encoding, and apply bioinformatics tools and scripts for feature engineering.
- Comprehend the fundamentals of representation learning and its applications in genomics, focusing on techniques such as word2vec, DNABERT2, and Nucleotide Transformer
- Apply machine learning pipelines for genomic sequence classification, utilizing both feature engineering and representation learning techniques, and gain hands-on experience with relevant tools and libraries.
- Evaluate and compare feature extraction and representation learning approaches in terms of their effectiveness for genomic sequence classification task.
Intended Audience and Level
This tutorial is designed for participants at different academic levels, from undergraduate students to early career researchers, as well as scientists from both academic and industry backgrounds, who have an interest in genomic sequence analysis and Machine Learning. We encourage participants to follow along with the hands-on exercises, whether they choose to actively participate or simply observe. For those who wish to engage in hands-on training, we recommend having a basic understanding of a few prerequisites: familiarity with running commands in a terminal, foundational knowledge of computer programming (preferably in Python), and a basic understanding of Machine Learning concepts.
The slides and materials for the hands-on exercises (scripts and datasets) will be shared online prior to the tutorial. All resources will be freely accessible to participants through a dedicated GitHub repository.
Schedule
| 14:00-14:45 |
Lecture: Exploring Traditional Feature Engineering in Genomics
- Overview: Introduction to machine learning for sequence classification and methods to extract numerical information from biological sequences
- Introduction to traditional feature extraction:
- Sequence composition: GC content, nucleotide composition, basic k-mer and accumulated nucleotide frequency
- Numerical mapping: Z-curve and one-hot encoding
- Example of public software packages: Seq2Feature, iFeature and iLearn
|
| 14:45-15:45 |
Hands-on: Feature Extraction in Practice — Crafting Descriptors for Genomic Analysis
- Practical exercises for extracting traditional genomic features
- Application of basic ML pipeline for sequence classification
- Comparative analysis of the effectiveness of different feature extraction approaches
|
| 15:45-16:00 |
Coffee Break |
| 16:00-16:45 |
Lecture: Decoding the Genomic Language — Embeddings and Representations for Genomic Sequences
- Introduction to representation learning for genomic sequences, highlighting their role in advancing genomic analysis
- Introduction to embeddings: Overview of word embeddings, with a focus on Word2Vec and its application to genomic sequences
- Foundation models in genomics: Overview of LLMs, focusing on DNABERT2 and Nucleotide Transformer for sequence representation learning
|
| 16:45-18:00 |
Hands-on: Embedding Genomic Data — From Word Embeddings to Large Language Models
- Practical exercises applying Word2vec and LLMs (DNABERT2 and Nucleotide Transformer) to extract features from genomic sequences
- Evaluation and comparison of the features obtained from traditional methods and embedding techniques
|
- top -
Tutorial VT1: Visualising and interpreting your -omics results using ggplot2 and R
Room: Virtual
Date: July 14, 2025
Start Time: 09:00
End Time: 18:00
Organizer:
Emily Johnson
Speakers:
Emily Johnson, University of Liverpool
Euan McDonnell, University of Liverpool
Lauren Mee, University of Liverpool
Max Participants: 30
Description
This full-day tutorial introduces participants to the principles of impactful data visualisation and equips them with the skills to create publication-ready visualisations for -omics data. Designed for beginners with basic knowledge of the R programming language, the tutorial will guide attendees through creating and interpreting key visualisations such as volcano plots, box plots, heatmaps, dot plots, and network diagrams.
Participants will work with real biological datasets in Quarto notebooks and learn how to use tools like ggplot2, ComplexHeatmap, igraph, ggraph, and ClusterProfiler. Through hands-on coding exercises and interactive lectures, attendees will develop an intuition for ggplot2’s grammar of graphics, best practices in data visualisation, and the application of functional enrichment methods to contextualise results.
Whether you are a student or researcher wanting to improve your visualisation skills, or a computational biologist looking to enhance the presentation of your results, this tutorial will provide the tools and knowledge to produce professional-quality figures.
Learning Objectives
- To understand the principles of good data visualisation, including best practices and common pitfalls to avoid.
- To learn about the most common types of visualisations for -omics data, including volcano plots, box plots, heatmaps, dot plots, and networks:
- Create each type of visualisation using appropriate R packages.
- Effectively interpret outputs generated.
- To develop a working understanding of ggplot2 syntax, enabling participants to apply learned concepts to their own datasets.
- To learn how to produce and interpret biological networks.
- To contextualise -omics results using functional enrichment techniques such as Over-Representation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA).
Intended Audience and Level
Beginner. This tutorial is suitable for students and researchers want to learn more about common omics visualisations and how to produce them, and computational biologists that might want to enhance their visualisations. Basic familiarity with R is recommended to get the most out of the tutorial. Prior exposure to omics data is beneficial but not mandatory for this tutorial.
Schedule
| 09:00-09:45 |
Lecture: Introduction to Data Visualization
- Welcome and introductions
- Overview of most common types of data visualisation in omics papers
- What makes for a good or bad data visualisation
- Interactive quiz – participants will be given the opportunity to apply knowledge gained and suggest how they’d improve various data visualisations
Emily Johnson |
| 09:45-10:30 |
Lecture: introduction to ggplot2
- Introduction to the tidyverse and why ggplot2 is ideal for reproducible visualisation
- Overview of the “grammar of graphics” and ggplot2 syntax
- Examples of advanced visualisations produced with ggplot2
- Live coding demo to introduce core concepts
Lauren Mee |
| 10:30-10:45 |
10:30 - 10:45: Coffee Break |
| 10:45-12:00 |
Hands-on: plotting omics data with ggplot2
A follow along tutorial covering volcano plots and box plots to explore a differential expression analysis. For each type of visualisation, we will start from the most basic way to produce the plot and iteratively build up the plot to create an intuition for ggplot2 syntax. Themes and colour palettes will also be introduced during this session. The differential expression analysis results will be pre-processed. |
| 12:00-13:00 |
12:00 - 13:00: Lunch Break |
| 13:00-13:45 |
Hands-on: creating heatmaps with ggplot2 and ComplexHeatMap
A follow along tutorial that builds on from the morning practical. Participants will first create heatmaps using ggplot2, then they will be introduced to a more advanced heatmap-specific package: ComplexHeatMap. The merits of both approaches will be compared. The participants will also be introduced to the idea of clustering their data for visualisation purposes. |
| 13:45-14:15 |
Lecture: networks and how to interpret them
- Introduction to networks in biological data visualisation and where they’re appropriate
- Understanding network-specific terminology
- Examples of networks (e.g., protein-protein interactions, co-expression networks)
- Brief overview of inputs for and challenges of network visualisation
Euan McDonnell |
| 14:15-15:30 |
Hands-on: creating network visualisations with igraph and ggraph
Hands-on coding session where attendees will use igraph and ggraph to create network diagrams. Attendees will learn about different network layouts, how to identify and emphasise certain network structures and how to use igraph outputs as an input for ggraph (a package which is built on ggplot2 and compatible with tidyverse workflows) – for high quality and high-resolution network visualisations. |
| 15:30-15:45 |
Coffee break |
| 15:45-16:15 |
Lecture: Contextualising Results with Functional Enrichment
- What is functional enrichment and how does it help us make sense of our results?
- Common databases for enrichment analysis (e.g., GO, KEGG)
- Differences between Over-Representation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA), including best practices for both (use of appropriate background for ORA and choice of ranking metrics for GSEA)
- Visualising enrichment results: dot plots, ridge plots, and network diagrams.
Emily Johnson |
| 16:15-17:30 |
Hands-on: ORA and GSEA practical
Hands-on coding session using the ClusterProfiler package to perform ORA and GSEA on the results of a differential expression analysis. Participants will produce dot plots and ridge plots and compare the outputs of the two approaches. We will cover best practices for ORA and GSEA. There will be additional supplementary content including how to carry out functional enrichment for non-model organisms and advanced analysis such as Gene set variation analysis (GSVA). |
| 17:30-18:00 |
Wrap-Up and Discussion
- Review of key concepts covered in the tutorial.
- Q&A session and discussion on applying concepts to attendees’ own research projects.
|
- top -
Tutorial VT2: OmicsViz: Interactive Visualization and ML for Omics Data
Room: Virtual
Date: July 15, 2025
Start Time: 09:00
End Time: 18:00
Organizer:
Ragothaman M. Yennamalli
Speakers:
Ragothaman M. Yennamalli, SASTRA Deemed to be University
Shashank Ravichandran, Incedo Inc
Megha Hegde, Kingston University London
Jean-Christophe Nebel, Kingston University London
Farzana Rahman, Kingston University London
Max Participants: 30
Description
Data Science and Machine Learning are intricately connected, particularly in computational biology. In a time when biological data is being produced on an unprecedented scale — encompassing genomic sequences, protein interactions, and metabolic pathways- meeting the demand has never been more crucial.
Data visualization plays a crucial role in biological data sciences since it allows the transformation of complex, often incomprehensible raw data into visual formats that are easier to understand and interpret. This allows biologists to recognize patterns, anomalies, and correlations that would otherwise be lost in the sheer volume of data. In addition, machine learning (ML) has brought about a revolution in the analysis of biological data. Exploiting extensive datasets, ML provides tools to model complex systems and generate predictions. Indeed, ML algorithms excel at uncovering subtle patterns in data, contributing to tasks like predicting protein structures, comprehending genetic variations and their implications for diseases, and even facilitating drug discovery by predicting molecular interactions.
The integration of data visualization and machine learning is particularly powerful. In particular, visualization may aid in interpreting machine learning models, allowing biologists to understand and trust their predictions. It could also help fine-tune these models by identifying outliers or anomalies in the data.
Due to its remarkable capability, there has been a surge in the development and application of tools that combine data visualization and machine learning in biology. Platforms that integrate these technologies enable biologists to conduct comprehensive analyses without needing deep expertise in computer science. Assuredly, this democratization of data science and ML has empowered more and more biologists to engage in sophisticated, data-driven research.
Learning Objectives
This tutorial is divided into two parts. In the first part of the tutorial, the participants will learn how to install
and use tools for data visualization using Python.
The second part will focus on installing and using ML tools for feature selection, model training, and model optimization using Python.
By the end of this tutorial, the participants will be able to:
- Explain the role and significance of data visualization in the context of scientific research. (Revised Bloom’s Taxonomy [RBT] Assessment: Understand)
- Apply fundamental principles of data visualization to create clear and informative visual representations of data. (RBT Assessment: Apply)
- Create a variety of data visualizations using Python libraries, i.e., Matplotlib, Seaborn, and Plotly. (RBT Assessment: Understand)
- Understand the basics of colour theory and its implications for creating accessible and aesthetically pleasing visualizations. (RBT Assessment: Understand)
- Design data visualizations that are accessible to a diverse audience, including those with colour vision deficiencies. (RBT Assessment: Create)
- Gain practical skills in preprocessing data and selecting appropriate features for machine learning models. (RBT Assessment: Apply)
- Build, train, and evaluate machine learning models using Python libraries like Scikit-learn and TensorFlow/Keras. (RBT Assessment: Analyze)
- Implement machine learning algorithms on real-world biological datasets, demonstrating an understanding of the application of these techniques in biology. (RBT Assessment: Apply)
- Create integrated visualizations of machine learning results using tools like Yellowbrick, Bokeh, and TensorBoard. (RBT Assessment: Create)
- Critically evaluate and discuss the applications, challenges, and implications of data visualization and machine learning in scientific research, particularly in biology. (RBT Assessment: Evaluate)
Intended Audience and Level
The tutorial is aimed towards participants at the level of Graduate students, researchers, and scientists in both
academia and industry who are interested in Data Visualization and ML. Prerequisites: Basic to intermediate
knowledge in Python and basic knowledge in machine learning.
Schedule
| 09:00-10:00 |
Lecture: Introduction to Data visualization: Importance and Basic principles of data visualization in scientific research
Jean-Christophe Nebel |
| 10:00-10:45 |
Hands-on: Python Libraries for Visualization: Matplotlib, Seaborn, Plotly and others
Farzana Rahman, Ragothaman Yennamalli, Shashank Ravichandran, and Megha Hegde |
| 10:45-11:00 |
10:45AM - Coffee/Tea Break |
| 11:00-12:00 |
Lecture: Colour theory in Visualization: Colour palettes, Accessible and Inclusive visualizations
Ragothaman Yennamalli |
| 12:00-13:00 |
Hands-on: Creating various types of charts, plots for clarity and aesthetics
Case studies with real world datasets
Farzana Rahman, Ragothaman Yennamalli, Shashank Ravichandran, and Megha Hegde |
| 13:00-14:00 |
Lunch Break |
| 14:00-15:00 |
Lecture: Fundamentals of Machine Learning: Types of ML, Data preprocessing and feature
selection, model selection and training
Ragothaman Yennamalli and Farzana Rahman |
| 15:00-16:00 |
Hands on: Python libraries for Machine Learning: Scikit-learn, Pandas, NumPy, TensorFlow/Keras Building models using real-world biological data
Shashank Ravichandran, and Megha Hegde |
| 16:00-16:15 |
Coffee/Tea Break |
| 16:15-17:15 |
Hands on: Integrating Data Viz and ML: Yellowbrick, Bokeh, Tensorboard, Scikit-plot, etc.
Farzana Rahman and Megha Hegde |
| 17:15-18:00 |
Question and Answer session
Identify and highlight blocks of hands-on content in your submission |
- top -
Tutorial VT3: Computational approaches for deciphering cell-cell communication from single-cell transcriptomics and spatial transcriptomics data SOLD OUT
Room: Virtual
Date: July 15, 2025
Start Time: 09:00
End Time: 13:00
Organizer:
Giulia Cesaro
Speakers:
Giulia Cesaro, University of Padova
James Shiniti Nagai, RWTH Aachen University
Mayra Ruiz, RWTH University Hospital
Barbara di Camillo, University of Padova
Ivan G. Costa, RWTH University Hospital
Giacomo Baruzzo, University of Padova
Max Participants: 40
Description
Tissues and organs are complex and highly-organized systems composed of diverse cells that work together to maintain homeostasis, drive development and mediate complex disease progression as Myocardial Infarction (Kuppe et al. 2022). A key focus of modern biology is understanding how heterogeneous populations of cells coexist and communicate with each other (intercellular signaling), how they properly respond (intracellular signaling) within a tissue and organ system and how these processes vary across different experimental conditions (comparative analysis). Recently, a rapid expansion of computational tools exploring the expression of ligand and receptor has enabled the systematic inference of cell-cell communication from single-cell transcriptomics and spatial transcriptomics data (Armingol et al. 2021; Armingol, Baghdassarian, and Lewis 2024). These are crucial in unravelling the complex landscape of biological systems.
This tutorial aims to provide a comprehensive introduction to computational approaches for cell-cell communication inference using high throughput transcriptomics data. It covers the fundamental concepts of cellular communication and assumptions underlying analysis focusing on the main computational methods used in the field. This includes computational approaches for inter-cellular communication inference (CellphoneDB (Efremova et al. 2020); LIANA (Dimitrov et al. 2022, 2024)) and for intra-cellular signals communication (scSeqComm (Baruzzo, Cesaro, and Di Camillo 2022); NicheNet (Browaeys, Saelens, and Saeys 2019)). Next, we will describe approaches for comparative analysis of cell-cell networks in distinct biological conditions (CrossTalkeR (Nagai et al. 2021)) and methods for spatially resolved cell-cell communications (Ischia (Regan-KomitoDaniel 2024); DeepCOLOR (Kojima et al. 2024)).
In the first part of the tutorial, participants will be introduced to the theoretical basis of state-of-the-art computational approaches and will learn how to use representative tools for inferring intercellular signaling and intracellular signaling pathways. In the second part, we will focus on the comparative analyses, i.e. changes of cell-cell communication in two conditions, and subsequently highlighting the unique insights spatial transcriptomics data can provide for understanding tissue architecture and cellular communication. Both sections will be followed by a hands-on component based on the analysis of single cell and spatial transcriptomics data from the myocardial infarction atlas (Kuppe et al. 2022). To promote transparency, all the codes, software tools and the datasets used throughout the tutorial will be available and accessible through open-access repositories (e.g. GitHub repositories or Zenodo platforms).
Learning Objectives
- Understanding and identifying the key theoretical concepts of cell-cell communication analysis
- Learn the foundations of main computational approaches for cell-cell communication inference and develop critical thinking skills to choose and apply the most appropriate tools tailored to the specific research questions and analysis contents
- Gain hands-on experience in applying these computational methods to real-world data and learn how to interpret and evaluate the results in the context of biological systems
Intended Audience and Level
This tutorial is aimed at Master’s or PhD students, as well as researchers in the fields of bioinformatics, computational biology, medical informatics, and related fields. It is designed for those who are users and/or developers of bioinformatics software and are interested in incorporating cell-cell communication analysis into their workflows. The tutorial is suitable for beginner to intermediate users of computational tools and those looking to expand their skill set in cell-cell communication analysis. Basic knowledge of R and Python is strongly recommended, as well as previous experience in gene expression and next-generation sequencing analysis.
Schedule
| 09:00-09:30 |
Introduction to computational inference of intercellular and intracellular cell-cell communication
Overview of cell-cell communication inference, assumptions and challenges, a priori biological knowledge, current approaches for intercellular and intracellular signaling
Di Camillo, Baruzzo |
| 09:30-10:30 |
Hands-on analysis of Myocardial Infarction scRNA-seq
Presentation of case study: human myocardial infarction, tutorial on cell-cell communication analysis from single cell transcriptomics data
Cesaro (Helpers: Baldan Matteo and Tussardi Gaia) |
| 10:30-10:45 |
Coffee Break |
| 10:45-11:15 |
Introduction to comparative analysis of cell-cell communication and spatial analysis
Overview of cell-cell communication comparative analysis and computational approaches, spatial transcriptomics technologies and platforms: pro and cons
Costa |
| 11:15-11:45 |
Hands on analysis of Myocardial Infarction ST and scRNA-seq (part 1)
Tutorial on comparative analysis from single-cell transcriptomics and spatial transcriptomics data using myocardial infarction case study
Nagai, Ruiz (Helpers: Thiago Maié and Kai Peng) |
| 11:45-12:00 |
Coffee Break |
| 12:00-12:45 |
Hands on analysis of Myocardial Infarction ST and scRNA-seq (part 2)
Tutorial on comparative analysis from single-cell transcriptomics and spatial transcriptomics data using myocardial infarction case study
Nagai, Ruiz (Helpers: Thiago Maié and Kai Peng) |
| 12:45-13:00 |
Q&A Conclusion |
- top -
Tutorial VT4: An applied genomics approach to crop breeding: A suite of tools for exploring natural and artificial diversity
Room: Virtual
Date: July 15, 2025
Start Time: 14:00
End Time: 18:00
Organizer:
Maria Skrabisova
Speakers:
Trupti Joshi, University of Missouri-Columbia
Yen On Chan, University of Missouri-Columbia
Maria Skrabisova, Palacky University in Olomouc
Jana Biova, Palacky University in Olomouc
Max Participants: 50
Description
The urgent need for crop improvement is hindered by the lack of precision in crop breeding. Although significant progress has been made in genomics, many causal genes of important agronomic and nutritional traits remain unknown. This is due to the inefficient identification of causal genetic features in candidate genes. With the growing body of sequencing data and phenotype information, current advances in genomics and the development of bioinformatics tools offer improvements in candidate gene selection. In this workshop, SoyHUB, the suite of tools, strategies, and solutions for soybean applied genomics, will be presented along with its extension to other crops at KBCommons. Our methodology for data integration, curation, reuse, and leveraging will be highlighted. Practical utilization of integrated data with the tools will be demonstrated. We will focus on the selection of best-performing markers, identification of causal genes, and exploration of alleles and genomic variation. We will cover simple Mendelian traits, showing how to analyze variation in protein-coding regions and promoters and touch on copy number variation. Solutions for complex cases, such as multiple independent alleles in a single gene or quantitative traits, will also be introduced. This workshop will highlight recent achievements in leveraging big data to improve precision in GWAS-driven discoveries and, therefore, accelerate the breeding of soybean and other crops.
Learning Objectives
- To understand the basics of fine-mapping QTLs by post-GWAS analyses
- To understand the importance of identifying causative mutations in causal genes
- To learn principles of curating resequenced data gained from independent studies
- To learn how to explore genetic variation
- To understand the specifics of the Allele Catalog, GenVarEx, AccuTool, AccuCalc, SNPViz v2.0 and MADis tools
- To gain hands-on experience in applying tools and interpreting results for soybean as a model by using a web-based suite of tools, the SoyHUB
Intended Audience and Level
Graduate students, researchers, scientists, and practitioners in both academia and industry who are interested in applications of bioinformatic tools for crop breeding improvement. The tutorial is aimed towards entry-level participants with knowledge of the fundamentals of biology and bioinformatics (beginner). No prior experience with Python programming language is assumed, but familiarity with working on Unix-based systems is strongly recommended for the participants.
The tutorial slides and materials for hands-on exercises (e.g., links to demo, code implementation, datasets) will be posted online prior to the tutorial and made available to all participants.
Schedule
| 14:00-14:45 |
Translational Bioinformatics Frameworks and AI Solutions for Multiomics Research
Next-generation sequencing and multiomics data (bulk and single-cell) capturing molecular changes from genomics all the way to phenomics, have become an integral part of research in all domains including biomedical sciences, plants sciences, and others. This rapid revolution in the multiomics has posed a growing need for translational tools that can handle large amounts of data, are easily expandable, provide interpretable results and can be readily applied to any species. To address such translational needs, we have developed Soybean Knowledge Base (SoyKB) and Knowledge Base Commons (KBCommons) web-based frameworks, both fully equipped to handle the entire multiomics landscape for all organisms. SoyHUB within SoyKB provides access to our developed suite of tools such as AccuTool, Allele Catalog, GenVarX, MADis and others, specifically designed to provide the plant community with efficient data driven solutions for better breeding strategies. Additionally, our G2PDeep, deep learning method, provides a comprehensive web-based resource for phenotype predictions using multiomics data for all organisms.
Trupti Joshi |
| 14:45-15:40 |
Diversity Panel Creation and Resequencing Data Curation Using SnakyVC Pipeline, Allele Catalog Pipeline, and Allele Catalog Tool (hands-on)
With the growing availability of large-scale genomic datasets, in silico identification of causal genes and crop improvement have become more achievable. However, extracting meaningful insights from these datasets often requires extensive data processing using various computational tools, which can be time-intensive due to sequential tool transitions. To address this challenge, we developed SnakyVC, a scalable variant calling pipeline to process large-scale whole-genome resequencing (WGRS) data, and an Allele Catalog Pipeline to annotate SNPs and Indels and generate Allele Catalog datasets. These high-efficiency and parallelizable pipelines can be deployed on standalone servers or high-performance computing clusters to significantly reduce computational time. To expand the benefits of the Allele Catalog datasets, the web-based Allele Catalog Tool has been developed and integrated into the SoyKB and KBCommons web platforms. This tool enables researchers to query and visualize alleles within genes, functional annotations, accession metadata, and phenotype distributions. Currently, this tool supports a wide range of organisms including soybean, Arabidopsis , poplar, rice, sorghum, and maize. With its extensive functionalities and new organism integration capabilities, the Allele Catalog Tool facilitates the discovery of novel alleles and the selection of plant accessions for improved breeding strategies and agricultural traits. Together, these tools empower researchers to efficiently process genomic data and enhance crop improvement efforts.
Yen On Chan |
| 15:40-16:00 |
Coffee break |
| 16:00-17:45 |
Utilization of the SoyHUB tools in Post-GWAS analyses (hands-on)
This hands-on tutorial will cover approaches and strategies in applied genomics that have been developed for Soybean and are available for adoption for other crops. Using SnakyVC, we created a panel of diverse resequenced soybean accessions. To explore the genetic diversity of Soy2939, we demonstrate the use of a gene-centric toolbox for post-GWAS analysis, SoyHUB. Specifically, we will present allele mining in protein-coding regions (Soybean Allele Catalog), gene regulatory regions (GeneVarEx), and intergenic regions (SNPViz v2.0). Additionally, we will introduce a post-GWAS strategy, the Synthetic Phenotype to Causative Mutation (SP2CM) approach, which increases the probability of identifying causative mutations. We will also demonstrate how to use AccuTool to calculate accuracy as a measure of the direct correspondence between phenotype and variant position. In this tutorial, we will perform demonstration analyses using each tool within the SoyHUB platform and discuss the results in the context of relevant biological insights. Together, the strategy and tools provide a powerful, bioinformatics-driven layer in the pre-breeding phase, that ultimately improves and accelerates crop breeding.
Maria Skrabisova |
| 17:45-18:30 |
MADis: Genomic Analysis Tool for the Revelation of Multiple Alleles within a Single Gene (hands-on)
Understanding crop diversification through evolution and domestication is crucial for crop breeding. Genome-wide association study (GWAS) has emerged as a powerful tool for mapping genomic loci linked to important traits. However, GWAS often struggles to resolve complex genetic architectures, here we focus on a frequent situation when multiple independent causative alleles exist within a single gene. To address this GWAS limitation, we developed the MADis (Multiple Allele Discovery) tool, an innovative tool that computes a score for a combination of variant positions in a single candidate gene and, based on the highest score, identifies the best number and combination of CMs. In this hands-on tutorial, we will introduce the MADis tool and cover its functionalities and utilization. Participants will explore how MADis overcomes the limitations of traditional GWAS to accelerate precision breeding and enhance the understanding of complex genetic traits. Participants will also learn how to integrate MADis into their research workflow. The tool is available as a Python package on GitHub and as a web-based Soybean MADis tool specifically designed for a curated panel of 1066 soybean resequenced accessions.
Jana Biova |
- top -
Tutorial VT5: Comprehensive Bioinformatics and Statistical Approaches for High-Throughput Sequencing Data Analysis, Including scRNA-seq, in Biomarker Discovery
Room: Virtual
Date: July 14, 2025
Start Time: 14:00
End Time: 18:00
Organizer:
Xiaoli Zhang
Speakers:
Xiaoli Zhang
Lianbo Yu
Max Participants: 50
Description
With the significant advancements in genomic profiling technologies and the emergence of selective molecular targeted therapies, biomarkers have played an increasingly pivotal role in both the prognosis and treatment of various diseases, most notably cancer. This workshop is designed to begin with an introductory overview of basic concepts of biomarkers, the diverse categories of biomarkers, commonly employed biotechnologies for biomarker detection, with a special focus on gene mutation and gene expression using DNA-seq, RNA-seq, and scRNA-seq data. Furthermore, we will discuss processes of biomarker discovery and development, and outlining the key steps involved and the current analytical methodologies utilized. Following this, we will discuss the identification of driver gene mutations and altered gene expression, using The Cancer Genome Atlas (TCGA) lung cancer data and PBMC scRNA-seq data as illustrative examples with using R code as practical demonstrations to enhance understanding. In the latter part of this workshop, we will discuss commonly utilized biostatistics and bioinformatics tools, including data visualization, survival analysis and machine learning methods, which are employed to predict disease progression and patient survival outcomes based on these critical biomarkers. By the conclusion of this course, participants will have acquired a broad and fundamental understanding of biomarker discovery, particularly in cancer. This encompasses key concepts, data sources, data analysis techniques, and interpretation strategies. Such expertise will equip participants with the knowledge necessary in contributing to the development of precision medicine in cancer patient treatment.
Learning Objectives
Through this tutorial, we expect that participants will have acquired a broad and fundamental understanding of biomarker discovery including key concepts, data types and sources, key steps in molecular biomarker discovery, commonly used biostatistics and bioinformatics methods in the field, and data analysis techniques for DNA-seq, RNA-seq, and scRNA-seq.
Intended Audience and Level
This introductory tutorial will be suitable for all bioinformaticians and biomedical researchers. R programming experience is preferred, but not required.
Schedule
| 14:00-14:45 |
Introduction of biomarkers
Definition of biomarkers, types of biomarkers, central dogma, molecular biomarkers, technologies for molecular biomarker detection, key steps in molecular biomarker discovery and related analytic methodologies involved. |
| 14:45-15:30 |
Identification of driver mutations
Discuss publicly available data sources, data types, use lung cancer TCGA DNA-seq and clinical data as examples to illustrate how to identify driver gene mutations in tumor samples. Hands on examples with R code for analysis will be demonstrated. |
| 15:30-15:45 |
Break |
| 15:45-16:45 |
Detection of differential expression
Statistical methods for analyzing differential expression in bulk RNA-seq and scRNA-seq data, complemented by visualization techniques such as t-SNE, UMAP, and heatmaps. Hands-on examples will be provided, including R code demonstrations for data analysis. |
| 16:45-17:00 |
Break |
| 17:00-18:00 |
Machine learning methods for phenotype prediction using biomarkers
Further discussing visualization of gene expression data with volcano plots, heatmaps, PCA plots, t-SNE, and UMAP etc., oncoplot and mutplot for gene mutation, logistic regression and survival analysis to predict biomarker association with patient progress and survival outcomes, then discuss some classification methods such as KNN, SVM, and random forest etc., for patient subtype analysis based on genomics data. Hands on examples with R code for predictive modeling will be demonstrated. |
- top -
Tutorial VT6: Beyond Bioinformatics: Snakemake for Versatile Computational Workflows
Room: Virtual
Date: July 14, 2025
Start Time: 09:00
End Time: 13:00
Organizer:
Carlos H. M. Rodrigues
Speakers:
Carlos H. M. Rodrigues, Australian Centre for Disease Preparedness
Max Participants: 20
Description
Snakemake is a powerful, Python-based workflow management system that revolutionises how computational tasks are designed, executed and reproduced. By allowing researchers to define workflows as a series of interconnected rules, Snakemake simplifies complex computational pipelines and ensures reproducibility across diverse scientific domains. This intensive workshop introduces participants to the full potential of Snakemake, moving beyond traditional bioinformatics applications to demonstrate its versatility in machine learning and data analysis.
Designed for researchers dealing with complex computational challenges, this tutorial will explore Snakemake’s capabilities through practical, hands-on examples that span multiple disciplines. Participants will learn how to create robust, scalable, and efficient workflows that can adapt to various research challenges. By the end of the workshop, attendees will have a comprehensive understanding of workflow management principles and the skills to implement sophisticated pipelines using Snakemake.
Learning Objectives
- Understand the fundamental concepts of workflow management with Snakemake
- Develop skills in creating flexible and reproducible computational pipelines
- Apply Snakemake to multiple domains, including bioinformatics and data science
- Implement advanced workflow features such as wildcards, parallel execution and configuration management
- Recognise best practices for designing efficient and maintainable workflows
Intended Audience and Level
This tutorial is designed for individuals across various disciplines who manage or analyse data: bioinformaticians, data scientists, computational researchers, and anyone wanting to know more about efficient data workflow management. Basic familiarity with command-line operations, such as executing scripts or simple commands like “ls” or “pwd”, will be beneficial for active participation. No prior Snakemake experience is necessary; we will cover everything from basics to advanced applications. For hands-sessions, participants will need a GitHub account to interact with the Binder environment where we will run the workshops. While live coding is encouraged, attendees are also welcome to observe and learn through demonstration if they prefer.
Schedule
| 09:00-09:30 |
Introduction to Snakemake and Workflow Principles |
| 09:30-10:45 |
Domain specific workflow examples
- Bioinformatics workflow (Hands-on)
- Data Science/Machine Learning workflow (Hands-on)
|
| 10:45-11:00 |
Coffee break |
| 11:00-12:15 |
Advanced Snakemake Features
- Wildcards and config files
- HPC Environments
- Parameterised simulation workflow (Hands-on)
|
| 12:15-12:45 |
- Practical considerations, Best Practices and Q&A Identify and highlight blocks of hands-on content
- Bioinformatics workflow (30-40 mins)
- Live coding of an RNA-seq data processing pipeline
- Participants create and modify Snakemake rules
- Explore rule dependencies and workflow structure
- Data Science/Machine Learning workflow (30-40 mins)
- Build an end-to-end ML pipeline
- Demonstrate data preprocessing, model training and evaluation
- Hands-on exercise in creating dynamic workflow rules
- Parameterised simulation workflow (30-40 mins)
- Create a parameterised simulation workflow
- Implement parameter sweeps
- Explore advanced features like wildcards and config files
|
- top -
Tutorial VT7: Assessing and Enhancing Digital Accessibility of Biological Data and Visualizations
Room: Virtual
Date: July 15, 2025
Start Time: 14:00
End Time: 18:00
Organizer:
Sehi L’Yi
Lawrence Weru
Thomas C. Smits
Nils Gehlenborg
Speakers:
Sehi L’Yi
Lawrence Weru
Thomas C. Smits
Nils Gehlenborg
Max Participants: 40
Description
As computational biologists, we produce biological datasets, visualizations, and computational tools. Our shared goal is to make our data and tools widely usable and accessible. However, we often fail to meet the needs of certain groups of people. Our recent comprehensive evaluation [1] (https://inscidar.org) shows that biological data resources are largely inaccessible to people with disabilities, with severe accessibility issues in almost 75% of all 3,112 data portals included in the study. To address the critical accessibility barriers, it is important to increase awareness of accessibility in the community and teach the workforce practical ways to enhance the accessibility of biological data resources. While there are existing resources and training opportunities that focus on content that includes no or little data, there is a lack of solutions and resources that provide insights into how to make data-intensive content accessible.
Our tutorial is designed to help participants understand the importance of digital accessibility in computational biology and practice various approaches to test and implement digital accessibility of biological data and visualizations. We will demonstrate our evaluation results [1] to help participants understand the critical barriers in biological research and education for people with disabilities, such as those involving vision, cognitive, and physical function. We will use hands-on examples that are familiar to and widely used by computational biologists, such as computational notebooks, genome browsers, and other visualizations. Our tutorial will conclude by introducing open problems and recent innovations, such as the accessibility of interactive genomics data visualizations [2]. We will ensure that our tutorial and all the materials are accessible.
Learning Objectives
- Understand the importance of accessibility and current accessibility problems in the field of computational biology.
- Learn different approaches to evaluate the accessibility of biological data resources.
- Learn approaches to enhance the accessibility of biological data resources and visualizations.
- Learn open problems and innovations in accessibility
Intended Audience and Level
Anyone in the computational biology workforce can benefit from this tutorial. The target audience includes (under)graduate students, postdocs, staff scientists, data analysts, software engineers, UI/UX designers, and principal investigators.
- This tutorial will be particularly helpful for people who create any form of digital content using biological data, such as visualizations, figures, websites, etc.
- For the hands-on sessions, experience in Python is helpful but not required. No additional programming experience is needed.
Schedule
| 14:00-15:00 |
Introduction & Background
● What is digital accessibility?
● What are accessibility techniques?
● Why is digital accessibility important in computational biology?
● What is the state of digital accessibility in computational biology?
● What can we do to improve it? |
| 15:00-15:30 |
Hands-on Session 1: Writing alt-text for biological data visualization
● Learn guidelines for writing alt text for biological data visualizations
● Practice writing alt text for biological data visualizations |
| 15:30-15:45 |
Coffee Break |
| 15:45-16:15 |
Hands-on Session 2: Keyboard accessibility of biological resources
● Learn the importance of keyboard accessibility in biological
websites
● Use the keyboard input only to navigate biological data resources |
| 16:15-16:45 |
Hands-on Session 3: Designing accessible biological visualization
● Learn accessibility guidelines for biological data visualizations
● Practice evaluating the accessibility of biological data
visualizations |
| 16:45-17:00 |
Coffee Break |
| 17:00-17:30 |
Hands-on Session 4: Making computational notebooks accessible
● Learn the importance of the accessibility of computational
notebooks
● Learn accessibility guidelines for computational notebooks
● Practice implementing the accessibility guidelines in computational
notebooks |
| 17:30-18:00 |
Open problems and innovations
● Making interactive biomedical data visualization accessible
● Complex data discovery tasks in data portals
● The INSCIDAR project (https://inscidar.org/)
● Useful resources |
- top -
Tutorial VT8: Generative AI for Single-Cell Perturbation Modeling: Theoretical and practical considerations SOLD OUT
Room: Virtual
Date: July 14, 2025
Start Time: 14:00
End Time: 18:00
Organizers:
Marina Esteban-Medina
Liya Zaygerman
Rosario Astaburuaga-García
Aspasia Orfanou
Vasileios Vasileiou
Speakers:
George Gavriilidis, Centre for Research and Technology Hellas
Konstantinos I. Giatras, National and Kapodistrian University of Athens
Gobikrishnan Subramaniam, Queen’s University Belfast
Sabrina Jagot, Institut NeuroMyoGen
Alejandro Madrid, Barcelona Supercomputing Center
Max Participants: 25
Description
Single-cell perturbation modelling is revolutionising our ability to predict how genes, drugs and external stimuli reshape cellular physiology. In this half-day virtual tutorial you will get hands-on with two highly performant Generative AI models—scGen and scPRAM—and learn how to benchmark them rigorously with the latest Perturb-Bench workflows. You will preprocess real single-cell datasets, train each model, evaluate them with metrics such as R² and Energy-distance, and discuss best practices for extrapolating to unseen patients and even across datasets. Practical notebooks, datasets and slides are distributed under a permissive CC-BY licence via GitHub and Jupyter Book (https://jupyterbook.org/en/stable/intro.html). Recorded versions of taught material will be available via YouTube after the conference.
Learning Objectives
- Understand single-cell perturbation modelling—how variational auto-encoders and attention-based optimal-transport models forecast unseen cellular responses.
- Implement scGen and scPRAM—from data normalisation through hyper-parameter tuning to interpretation of model outputs.
- Design biologically realistic experiments—including cross-study transfer and unseen-patient prediction scenarios.
- Benchmark models systematically—use Perturb-Bench pipelines, Optuna-powered hyper-parameter search and multiple evaluation metrics.
Intended Audience and LevelPost-graduate students and researchers comfortable with basic single-cell analysis and Python who wish to apply generative AI to perturbation biology, drug repurposing and computational modelling.
Data used for tutorial examples will be publicly available single-cell transcriptomics datasets made accessible via the pertpy package (https://pertpy.readthedocs.io/en/latest/usage/usage.html) and the perturbase repository (http://www.perturbase.cn/); also, datasets from the scPRAM publication will be used (https://doi.org/10.1093/bioinformatics/btae265). All of the data can also be found here: https://zenodo.org/records/15745452
Schedule
| 14:00-14:10 |
Title: Welcome and Introduction to perturbation modelling for single-cell technologies
- Short presentation (importance of perturbation modelling in single-cell biology, brief introduction to available tools, perturbation single-cell data, overview of workshop’s agenda)
Speaker: George Gavriilidis |
| 14:10-15:30 |
Title: scGEN: a landmark generative model for unseen perturbations
- Short presentation (Using autoencoders for manifold learning manifold learning, extrapolation to unseen events in single-cell perturbation data, scGEN architecture, possible real-world scenarios for deployment )
- Hands-on practical: (an enhanced version of https://pertpy.readthedocs.io/en/latest/tutorials/notebooks/scgen_perturbation_prediction.html will be designed focusing on single-cell data pre-processing, designating training and testing sub-datasets, model hyper-parameter tuning (epochs, batch size), running scGEN, interpreting model outputs, using dimensionality reduction to evaluate perturbational extrapolations, implementing basic metric R2 to monitor the accuracy of perturbation prediction for differentially expressed genes and highly variable genes)
Speaker: Konstantinos I. Giatras
Trainers: Gobikrishnan Subramaniam, Konstantinos I. Giatras |
| 15:30-15:45 |
Coffee Break |
| 15:45-16:45 |
Title: scPRAM: a newer perturbation generative model based on attention mechanism and causal counterfactuals
- Short presentation (Explore optimal transport principles in single-cell perturbation modelling, how scPRAM maps perturbations probabilistically between control and stimulated conditions, and its key components, including transport cost functions, regularisation, and interpretable metrics.)
- Hands-on practical (an enhanced version of https://github.com/jiang-q19/scPRAM/blob/main/Tutorial/PBMC_cross_celltype_predict.ipynb will be designed to focus on preprocessing single-cell datasets, configuring scPRAM with key hyperparameters(noise robustness, learning rate, batch size, and optimal transport settings), evaluating model performance using R2, Wasserstein distance and dimensionality reduction and showcasing how attention prioritises the most relevant cells or features for predicting perturbation responses.
Speaker: Sabrina Jagot
Trainers: Sabrina Jagot, George Gavriilidis |
| 16:45-17:00 |
11:45-12:00 AM Coffee Break |
| 17:00-17:50 |
Title: Decentralised benchmarking of generative perturbation models
- Short presentation (why benchmarking is important, selecting metrics beyond typical linear correlations, workflows and decentralised deployment)
- Hands-on practical (scGEN vs scPRAM; based on code from the ongoing Perturb-Bench effort from EU BH 2024 to systematically benchmark scGEN vs scPRAM against more metrics like E-distance, Maximum mean discrepancy in challenging single-cell scenarios like extrapolation to unseen patient perturbation responses and perturbation predictions across-species)
Speaker: Alejandro Madrid
Trainers: Alejandro Madrid, Konstantinos I. Giatras |
| 17:50-18:00 |
Title: Wrap up and discussion
- Short presentation (Short recap of key take away messages)
- Open discussion
Speaker: All |
- top -
Tutorial VT9: Biomedical text mining for knowledge extraction
Room: Virtual
Date: July 15, 2025
Start Time: 09:00
End Time: 13:00
Organizer:
Jake Lever
Speakers:
Jake Lever, University of Glasgow
Zaiqiao Meng, University of Glasgow
Javier Sanz-Cruzado Puig, University of Glasgow
Max Participants: 60
Description
Modern bioinformatics analyses rely heavily on the existing knowledge of the role of genes and mutations in different diseases, as well as the complex interactions between genes, proteins and drugs. However, access to this information is often limited for many biomedical problems, especially niche areas, due to lack of knowledge bases and large curation costs. The information is often locked in the text of the original research papers. Machine learning methods, particular natural language processing techniques, offer an automated approach to extracting knowledge from the research literature to build bespoke knowledge bases for scientists’ needs. This tutorial will provide a hands-on introduction to the core tasks in biomedical natural language processing (BioNLP). These include identifying mentions of important concepts (e.g. phenotypes, cell-lines, etc) and extracting nuanced relationships between them. Finally, it will show how large language models have changed how information can be quickly extracted, but also highlight their challenges.
The tutorial materials are available at https://ai4biomed.org/ismb2025tutorial/
Learning Objectives
- Identify different sources of biomedical text for use in text mining
- Implement methods for extracting mentions of biomedical entities (e.g. drugs and genes) from biomedical text
- Demonstrate approaches to extracting relationships between entities in biomedical text
- Apply large language models to various information extraction tasks
- Appraise the abilities of large language models for different natural language processing tasks
- Discuss approaches for evaluating the performance and quality of extraction methods
Intended Audience and Level
Bioinformatics researchers with an interest in extracting knowledge from text, particularly for knowledge base construction. They should be comfortable with coding in Python. No prior knowledge about machine learning or deep learning is expected.
Schedule
| 09:00-09:15 |
Introduction to Biomedical Natural Language Processing
This introduction will cover the applications of biomedical text mining and outlining the plan for the day. |
| 09:15-09:30 |
Lecture: Getting started in NLP
This talk will introduce the core concepts of getting computers to work with text, getting access to appropriate biomedical text and annotating it for building and evaluating text mining systems. |
| 09:30-10:00 |
Hands-on: Getting started in NLP
This session will have attendees try out some introductory NLP tasks in Python and get access to biomedical text from PubMed through its API |
| 10:00-10:15 |
Lecture: Identifying mentions of biomedical concepts using named entity recognition (NER)
This talk will go over the NER task and some of the different approaches. It will focus on transformer-based methods. |
| 10:15-10:45 |
Hands-on: NER with Spacy and HuggingFace
This session will have attendees work through a Jupyter notebook to train an NER system given a provided set of annotations. They will get to work with transformer-based models using the HuggingFace library. |
| 10:45-11:00 |
Coffee Break |
| 11:00-11:15 |
Lecture: Extracting relations from biomedical text
This talk will outline the importance of extracting meaningful relations between entities and going beyond co-occurrences. |
| 11:15-12:00 |
Hands-On: Relation Extraction with Co-occurrences and HuggingFace
This session will have attendees work through a notebook using data that has entities already extracted to extract associations. |
| 12:00-12:15 |
Lecture: Using Large Language Models for Biomedical Text Mining
This talk will focus on the strengths and weaknesses of large language models for the tasks discussed in this tutorial. It will give a brief background on how they work and the common pitfalls when used for information extraction |
| 12:15-12:45 |
Hands-On: LLMs for Entity Extraction and Relation Extraction
This session will have attendees work with a small LLM and apply it to pre-prepared data for several extraction tasks. |
| 12:45-13:00 |
Lecture: LLMs and the future of information extraction
The final talk will lead a discussion on the benefits of LLMs for information extraction, but also what challenges remain. |
- top -



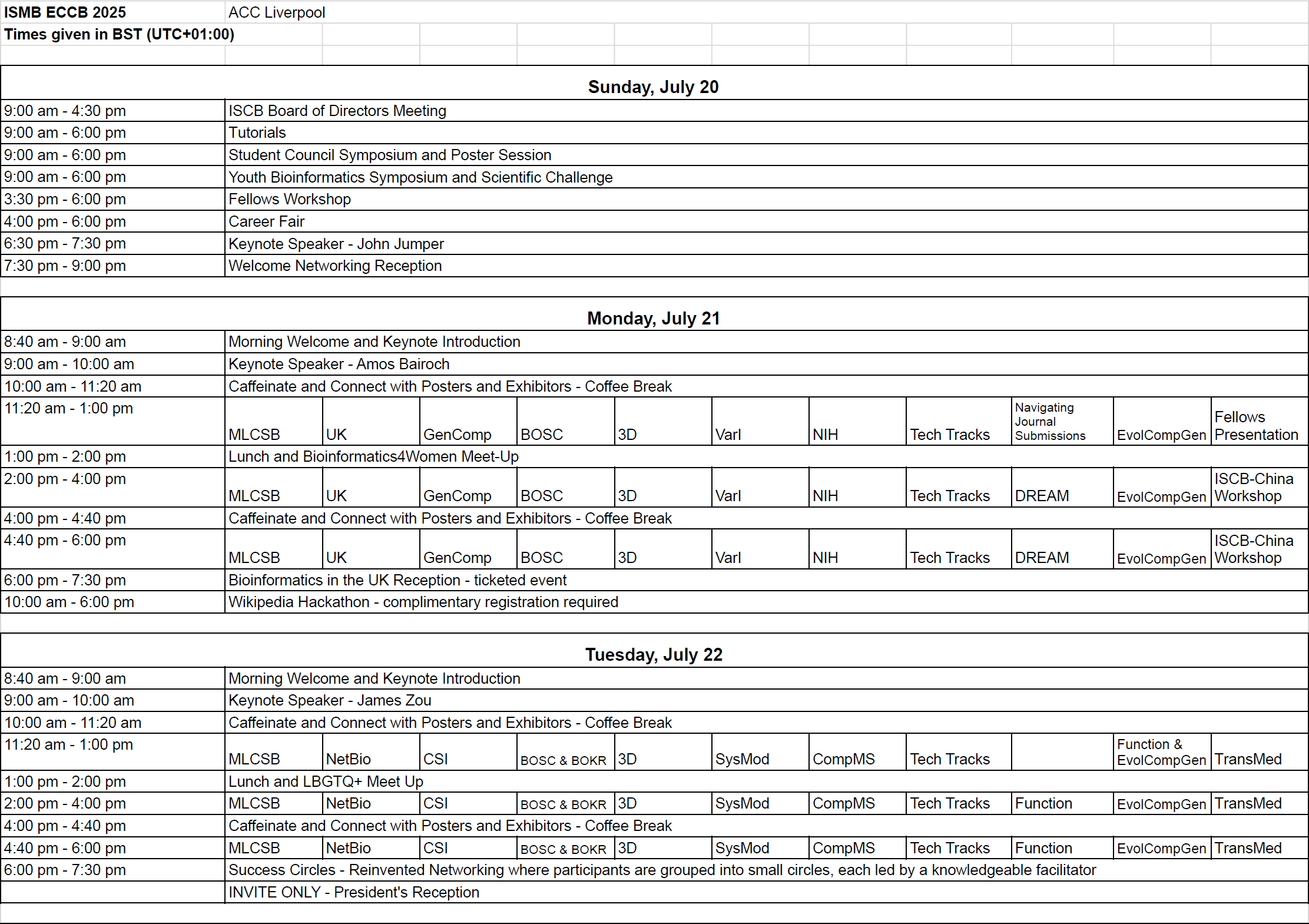

 It is impossible to fully understand biological systems without understanding the 3D structure of their constituting parts and their interactions. As such the topics relevant for 3DSIG are wide and include, but are not restricted to Structure-based drug discovery including polypharmacology and network pharmacology; Structure representation, classification and prediction;
It is impossible to fully understand biological systems without understanding the 3D structure of their constituting parts and their interactions. As such the topics relevant for 3DSIG are wide and include, but are not restricted to Structure-based drug discovery including polypharmacology and network pharmacology; Structure representation, classification and prediction;




 The large, complex data sets for the Critical Assessment of Massive Data Analysis (CAMDA) contest include built-in truths for calibration. In an open-ended competition, however, both seasoned researchers and cunning students push the boundaries of our field, with unexpected questions or angles of approach often bringing the most impressive advances.
The large, complex data sets for the Critical Assessment of Massive Data Analysis (CAMDA) contest include built-in truths for calibration. In an open-ended competition, however, both seasoned researchers and cunning students push the boundaries of our field, with unexpected questions or angles of approach often bringing the most impressive advances.
 The CompMS group promotes the efficient, high quality analysis of mass spectrometry data. The CompMS initiative covers various computational mass spectrometry application domains, including proteomics, metabolomics, and lipidomics.
The CompMS group promotes the efficient, high quality analysis of mass spectrometry data. The CompMS initiative covers various computational mass spectrometry application domains, including proteomics, metabolomics, and lipidomics.
 Education-COSI focuses on bioinformatics and computational biology education and training across the life sciences.
Education-COSI focuses on bioinformatics and computational biology education and training across the life sciences.
 Evolution and comparative genomics are deeply intertwined with computational biology. Computational evolutionary methods, such as phylogenetic inference methods or multiple sequence alignment are widely used, yet remain far from “solved” and are indeed intense areas of research.
Evolution and comparative genomics are deeply intertwined with computational biology. Computational evolutionary methods, such as phylogenetic inference methods or multiple sequence alignment are widely used, yet remain far from “solved” and are indeed intense areas of research.
 The mission of the Function Community of Special Interest (Function-COSI) is to bring together computational biologists, experimental biologists, biocurators, and others who are dealing with the important problem of gene and gene product function prediction, to share ideas and create collaborations.
The mission of the Function Community of Special Interest (Function-COSI) is to bring together computational biologists, experimental biologists, biocurators, and others who are dealing with the important problem of gene and gene product function prediction, to share ideas and create collaborations.

 iRNA track covers the full range of research topics in the field of RNA Biology, from computational and high-throughput experimental methods development to their application in different aspects of RNA processing, structure, and function.
iRNA track covers the full range of research topics in the field of RNA Biology, from computational and high-throughput experimental methods development to their application in different aspects of RNA processing, structure, and function.


 Regulatory genomics involves the study of the genomic control system, which determines how, when and where to activate the blueprint encoded in the genome. Regulatory genomics is the topic of much research activity worldwide. Since computational methods are important in the study of gene regulation, the RegSys COSI meeting focuses on bioinformatics for regulatory genomics.
Regulatory genomics involves the study of the genomic control system, which determines how, when and where to activate the blueprint encoded in the genome. Regulatory genomics is the topic of much research activity worldwide. Since computational methods are important in the study of gene regulation, the RegSys COSI meeting focuses on bioinformatics for regulatory genomics.


 The VarI COSI meeting is dedicated to the recent advances in the analysis and interpretation of the genetic variants.
The VarI COSI meeting is dedicated to the recent advances in the analysis and interpretation of the genetic variants.

