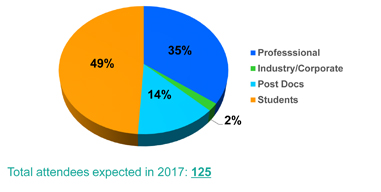
ISCB Africa ASBCB Conference on Bioinformatics 2017
SESSION 4: Bioinformatics of human genetics and population studies
Oral Presentation Abstracts
Insilico Identification of Protein-Coding and Non-Coding Regions in Next-Generation Technology Transcriptome Sequence Data: A Machine Learning Approach
Presenter:
Olaitan Awe
University of Ibadan
Additional authors:
Angela Makolo
University of Ibadan
Segun Fatumo
Wellcome Trust Sanger Institute
Wellcome Genome Campus, Hinxton, Cambridge
With the rapid increase in the volume of sequence data and multi-species transcripts generated using next-generation sequencing technologies, designing algorithms to process these data in an efficient manner and gaining biological insight is becoming a significantly growing challenge as there is no known effective method to discriminate between non-coding and protein-coding regions in human transcriptomes because RiboNucleic Acids (RNA) show similar features to each other. The few existing techniques mostly involve intense computation or multi-threading for small/large datasets to achieve small performance difference, and risking a high execution time of the tool.<br><br>To solve this problem, we developed a fast, accurate and robust alignment-free predictor based on multiple feature groups using Logistic Regression, for the discrimination of protein-coding regions in multispecies transcriptome sequence data, where the predictive performance is influenced by Open Reading Frame(ORF)-Related and ORF-unrelated features used in the model rather than the training datasets, thereby achieving a relatively high performance and computational speed in processing small and large datasets of full-length and partial-length protein-coding and non-coding transcripts derived from transcriptome sequencing.
We describe a series of experiments on the human datasets with a goal of generally performing better than competing techniques.
Our tool identified coding and non-coding regions in the human RNA-seq dataset with 97% accuracy, 97% F1-score, 97% sensitivity and 97% specificity.
We expect this new approach to result in an efficient computational cost of analyzing transcripts, and hence make genome annotation and transcriptome analysis easier.
Gene Regulatory Network Sparseness with Fuzzified Adjusted Rand Index (FARI)
Presenter:
Taiwo Adigun
University of Ibadan
Additional authors:
Angela Makolo
University of Ibadan Computer Science
Segun Fatumo
H3Africa Bioinformatics Network (H3ABioNet) Node, National Biotechnology Development Agency (NABDA), Federal Ministry of Science and Technology (FMST), Abuja
Gene regulatory networks have an important role in every process of life, including cell differentiation, metabolism, the cell cycle and signal transduction. Sparseness property of models and curse of dimensionality property of input data pose a serious challenge to its modeling. Besides, most developed network inference models did not model the effect of less expressed genes. We have proposed a modified technique called Fuzzified Adjusted Rand Index (FARI) to effectively handle the sparseness of gene regulatory network and to drastically reduce the effect of curse of dimensionality of the input data. The concept of Fuzzy is incorporated to calculate the sizes of intersection of expression values in the samples of the gene objects. An estimated fuzzified contingency table of two gene expression profiles from input dataset is generated and the ARI value of the two genes is calculated iteratively. We finally generated a distance matrix containing ARI values of all the genes in the dataset. The set of genes with higher values and the set of genes with lower values on each row (gene node) are separately fed into Recurrent Neural Network as parameters to train the learned model and to investigate the regulatory effect of least co-expressed genes on each gene. We have provided a fast and effective regularization technique to model a higher order neural network to handle large-scale biological network and to investigate the effect of less co-expressed genes on other genes.
COMBAT TB, an integrated environment for TB sequence data storage, analysis and visualization
Presenter:
Peter van Heusden
South African National Bioinformatics Institute, University of the Western Cape
Additional authors:
Thoba Lose
South African National Bioinformatics Institute
Ziphozakhe Mashologu
South African National Bioinformatics Institute
Alan Christoffels
South African National Bioinformatics Institute
Tuberculosis (TB), caused by the M. tuberculosis (Mtb) bacteria, continues to be one of the leading causes of morbidity and mortality in sub-Saharan Africa. The advent of Next Generation Sequencing (NGS) has allowed low cost sequencing of the 4 megabase Mtb genome and rapid growth of available sequence data. Availability of genomic sequence requires scalable data storage and analytic capacity to translate into knowledge for both public health laboratories and TB researchers. Under the auspices of the COMBAT TB project SANBI has developed an integrated environment for genomic data storage integrated with automated analysis workflows. This allows sequence data to be associated with metadata that is later available to enhance analysis. The integrated environment allows both “first pass” analysis of new sequence data using pre-built workflows and “ad-hoc” analysis of data as research needs dictate, using the Galaxy and COMBAT TB Explorer environments. The COMBAT TB Explorer (CTBE) is a graph database and visualisation environment that allows in-house data to be interpreted in the context of publicly available annotation of M. tuberculosis. The CTBE is built on a Neo4j graph database, using a data model inspired by the Chado and Global Alliance for Genomic Health (GA4GH) schemas for genomic and sequence variant annotation respectively. A task queue allows work to be shared between the CTBE and our analysis workflow environment. The complete COMBAT TB environment is available as a set of Docker containers, with all code available on Github.
Ensemble Clustering Algorithms: An Experimental Evaluation on Gene Expression Data
Presenter:
Itunuoluwa Isewon
Covenant University
Additional authors:
Faridah Ameh
Covenant University Department of Computer & Information Sciences
Efosa Uwoghiren
Covenant University Department of Computer & Information Sciences
Olufemi Aromolaran
Covenant University Department of Computer & Information Sciences
Jelili Oyelade
Covenant University Department of Computer & Information Sciences & Covenant University Bioinformatics Research (CUBRe)
Ensemble clustering provides more flexibility as the user is not constrained to the choice of a single clustering algorithm but can harness the strengths of multiple clustering approaches into one clustering solution and also avoid the possibility of making a poor choice of clustering algorithm. Ensemble clustering algorithms combine results produced by different clustering techniques through a consensus function. This study evaluated the performance of different consensus functions on five differently shaped or structured gene expression data sets. Four clustering algorithms were used to generate different clustering results from these data sets. These algorithms include: Fuzzy C-means algorithm, Spherical K-means algorithm, Complete-linkage Agglomerative Hierarchical clustering algorithm and Self Organising Maps algorithm. The base clusterings were combined in a cluster ensemble and re-clustered using the following consensus functions: Hard least square Euclidean consensus function, Soft least squares Euclidean consensus function, DWH (Dimitriadou, Weingessel and Hornik) consensus function, 3rd model of Gordon and Vichi consensus function, Soft median Manhattan consensus function. In order to perform a comparative analysis on the final clustering results gotten from these ensemble methods, the following internal cluster validity measures were used; Silhouette Width index, Connectivity Index and the Dunn Index. The results of the performance evaluation showed that the Soft median Manhattan consensus function was the best. Furthermore, the consensus clusters generated were insensitive to miss-classification from the individual clustering algorithms.
Reproducible bioinformatics workflows for heterogeneous African computational environments
Presenter:
Mamana Mbiyavanga
University of Cape Town
Additional authors:
Mustafa Alghali
University of Khartoum
Don Armstrong
University of Illinois
Shaun Aron
University of the Witwatersrand
Shakuntala Baichoo
University of Mauritius
Hocine Bendou
University of the Western Cape
Eugene de Beste
University of the Western Cape
Scott Hazelhust
University of the Witwatersrand
Fourie Joubert
University of Pretoria
Brian O'Connor
UCSC Genomics Institute
Souiai Oussama
Institut Pasteur de Tunis
Sumir Panji
University of Cape Town
Alex Rodriguez
University of Chicago
Yassine Souilmi
Mohammed V University, Rabat
Peter van Heusden
University of the Western Cape
Yi Long
University of the Western Cape
Azza Ahmed
University of Khartoum/Future University
Ayton Meintjes
University of Cape Town
Abayomi Mosaku
Covenant University
Phelelani Mpangase
University of the Witwatersrand
Lerato Magosi
University of Oxford
Nicky Mulder
University of Cape Town
Milt Epstein
University of Illinois
Victor Jongeneel
University of Illinois
Liudmila Mainzer
University of Illinois
Jenny Zermeno
University of Illinois
Gerrit Botha
University of Cape Town
Michael Crusoe
Common Workflow Language Project Co-founder, IT
Designing a bioinformatics pipeline that is runnable on any environment is challenging. Many things need to be taken into consideration: the setup of the software stack, the integration with the scheduler, how to control access to data, the tuning of parameters based on resources available and defining data locations for archival, processing, publishing and reference databases. H3ABioNet is responsible for supporting data analysis for H3Africa projects. It was therefore necessary to create SOPs to assist in the analysis process. We identified that these SOPs should be built into bioinformatics pipelines but also decided to containerise the software stacks involved, in order to reduce some of the aforementioned challenges. In August 2016 a five day hackathon was held by H3ABioNet where pipelines were developed and containerised for human exome variant calling, GWAS, SNP imputation and 16S rRNA analysis. The hackathon was an immense success and we managed to produce working code and good relationship between members. Post hackathon we spent time cleaning up our code and containers. Two of the pipelines were built using Nextflow while the other two made use of CWL. We used Docker to containerise the software stacks. All the code is available on GitHub and the containers have been deposited on Quay. Our future plans are to package our workflows using Singularity containers. Additionally since the packages make the process of software installation easier we plan to include these workflows as base material of some of the H3ABioNet training modules.
Enabling the processing of bioinformatic workflows where data is located through the use of cloud and container technologies
Presenter:
Eugene De Beste, University of the Western Cape, South Africa
Additional authors:
Alan Christoffels University of the Western Cape, South Africa
Antoine Bagula, University of the Western Cape, South Africa
he use of “big data” to inform biomedical decisions poses complex problems of storage, privacy and data security. This is especially true for fields such as e-health which deal with human health records. Organisations holding such data need to be able to assure regulators and patients of the security of their data storage and handling. In addition, when dealing with large datasets, movement of data for processing can pose a challenge. Many applications that are used to process various types of data have strict software package dependencies, imposing competing requirements on the administrators of institutional computing platforms. Software containers are a lightweight and generally better performing, albeit less diverse alternative to virtual machines. The advancement and increase in adoption of these container technologies have resulted in adaption for use in a variety of scenarios and fields. This allows researchers to replace the shipment of data with shipment of code by packaging their software into containers. Researchers are allowed to define their own toolchains and workflows to do analysis with rather than being limited to what has been allowed by the organisation managing the data set. Utilizing the growing cloud environment ecosystem, with platforms such as OpenStack, it is possible to provide researchers with an easy to use interface to execute custom workflows remotely, without the hassle of software dependency management and direct technical knowledge and reducing the need to send potentially large data sets from one location to another.
- top -
ISCB Africa ASBCB Conference on Bioinformatics 2017
SESSION 3: Bioinformatics of human genetics and population studies
Oral Presentation Abstracts
A bioinformatics protocol for analyzing cancer patient survival based on transcriptome datasets
Presenter:
Gaston Mazandu
African Institute for Mathematical Sciences
Additional authors:
Samuel Mensah
African Institute for Mathematical Sciences
Mathematical Sciences
Andrews Frimpong
African Institute for Mathematical Sciences
Mathematical Sciences
Chinenye Nworah
African Institute for Mathematical Sciences
Mathematical Sciences
Alexis Nshimiyimna
African Institute for Mathematical Sciences
Mathematical Sciences
Simukai Utete
African Institute for Mathematical Sciences
Mathematical Sciences
Emile Chimusa
University of Cape Town
Pathology
Nicola Mulder
University of Cape Town
Integrative Biomedical Sciences
According to the World Health Organization (WHO), cancer is currently a leading cause of death worldwide, with an estimate of 8.8 million cancer-related deaths in 2015. Cancer stem cells are regulated by complex interactions with the components of the tumor micro-environment through networks of cytokines and growth factors. These interactions are mediated by a group of proteins and microRNAs (miRs), which are expressed or repressed. These expression levels are critical for cancer stem cell formation and expansion, enabling the promotion of tumor cell proliferation and migration, as well as for the survival of cancer recurrence and patient survival. Microarray and RNA-seq provide tools to generate genome wide transcriptome information, deciphering global gene expression patterns and quantifying a large dynamic range of expression levels. In this study, we use datasets from The Cancer Genome Atlas (TCGA) to develop an expression level-based model to: (1) Predict the breast cancer patient survival, and (2) Identify genes specific to cancer patients and critical to their survival. This new pipeline has identified SIN3A, CSNK2A1, FBXO22, SLC44A4, LPCAT3, ITGAX and SLC22A23 as breast cancer associated genes of which SLC44A4 and ITGAX were previously reported to be related to cancer. Furthermore, we noticed that patients who expressed high levels of SLC44A4 and low levels of ITGAX were likely to survive longer compared to patients with low levels of SLC44A4 and higher levels of ITGAX. This may contribute to the detection of cancer case and the development of effective therapeutic strategies.
A multi-scenario genome-wide medical population genetics simulation framework (FractalSIM)
Presenter:
Jacquiline Mugo
University of Cape Town
Additional authors:
Ephifania Geza
Computational Biology Division, Institute of Infectious Disease and Molecular Medicine, University of Cape Town and African Institute for Mathematical Sciences, Muizenberg, Cape Town Department of Integrative Biomedical Sciences
Gaston Mazandu
Computational Biology Division, Institute of Infectious Disease and Molecular Medicine, University of Cape Town and African Institute for Mathematical Sciences, Muizenberg, Cape Town
Department of Integrative Biomedical Sciences
Joel Defo
Computational Biology Division, Institute of Infectious Disease and Molecular Medicine, University of Cape Town
Department of Integrative Biomedical Sciences
Samar Elsheikh
Computational Biology Division, Institute of Infectious Disease and Molecular Medicine, University of Cape Town
Department of Integrative Biomedical Sciences
Nicola Mulder
Computational Biology Division, Institute of Infectious Disease and Molecular Medicine, University of Cape Town
Department of Integrative Biomedical Sciences
Emile Chimusa
Division of Human Genetics, Faculty of Health Sciences, Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Observatory, Cape Town
Department of Pathology za:
Recent technological advances in high-throughput sequencing and genotyping have facilitated an improved understanding of genomic structure and disease-associated genetic factors. In this context, simulation models can play a critical role in revealing various evolutionary and demographic effects on genomic variation, enabling researchers to assess existing and design novel analytical approaches. Although various simulation frameworks have been suggested, they do not account for natural selection and genomic variation affecting diseases in admixture processes using resampling approaches (known to be fast and effective in mimicking real datasets), they are tailored to a single chromosome or a genomic region, very few capture large-scale genomic data, and are not accessible for genomic communities. We have developed a multi-scenario genome-wide medical population genetics simulation framework that has the capability to accurately mimic and generate genome-wide data under various genetic models on genetic diversity, genomic variation affecting diseases and DNA sequence patterns of admixed and/or homogeneous populations. Moreover, the framework accounts for natural selection in both homogeneous and admixture processes. We have assessed the output using popular tools, and the results demonstrated its capability to accurately mimic real scenarios and can be used to evaluate the performance of a range of genomic tools from ancestry inference to genome-wide association studies. This research work was published in June 2017.
A genome-wide association study of cardiometabolic traits in a sub-Saharan Africa population
Presenter:
Segun Fatumo
Wellcome Trust Sanger Institute
Cardiometabolic risk factors and its metabolic consequences of hypertension, type 2 diabetes, cardiovascular disease, and other co-morbidities continue to represents one of the biggest health challenges facing the world today and their prevalence is rising in Sub-Saharan Africa (SSA). Genome-wide association studies (GWAS) have identified novel genetic loci associated with a wide range of cardiometabolic traits, however a paucity of them has to date focused on SSA. We present the results of a GWAS comprising 34 traits and low coverage whole genome sequence (WGS) and dense array data from 1622 and 4778 individuals, respectively, from the MRC/UVRI General Population Cohort (GPC) study in rural south-west Uganda. . We identify a novel locus associated with HbA1C levels in the HbA2 gene on chr16 (P=6.9e-19). This 3.8kb deletion is common among Africans (MAF=0.25) and rare among Europeans (MAF=0.005), and has been previously shown to be strongly associated with several haematological traits among African-Americans—consistent with our findings from the GPC. We also identify and replicate a novel association signal specific to African populations at the GPT locus with alanine transaminase. In addition, we identify a novel association with serum bilirubin on the X chromosome for variant rs146474788, which is unique to African populations. Our findings provide proof-of-concept that in addition to providing an invaluable resource to researchers worldwide, such large-scale efforts in Africa can identify novel susceptibility loci associated with complex disease traits.
Designing the H3Africa genotyping array
Presenter:
Ananyo Choudhury
University of Witwatersrand
Additional authors:
Gerrit Botha
University of Cape Town
Computational Biology Division
Department of Integrative Biomedical Sciences
Emilson Chimusa
University of Cape Town
Division of Human Genetics
Scott Hazelhurst
University of Witwatersrand
School of Electrical & Information Engineering
Mamana Mbiyavanga
University of Capetown
Computational Biology Division
Department of Integrative Biomedical Sciences
Ayton Meintjes
University of Capetown
Computational Biology Division
Department of Integrative Biomedical Sciences
Adebowale Adeyemo
National Institutes of Health USA
National Human Genome Research Institute
Zané Lombard
National Health Laboratory Service & University of the Witwatersrand-Division of Human Genetics
Nicola Mulder
Computational Biology Division, IDM
University of Cape Town
Dept Integrative Biomedical Sciences
Despite a significant decrease in the cost of whole genome sequencing (WGS) in the recent years, single nucleotide variant (SNV) genotyping arrays remain the stalwart of genetic association studies. This is because of cost-effectiveness as well as the ease of data storage and analysis. The arrays currently used for population-genetic and genome-wide association studies in African populations, however, predominantly represent common Eurasian genetic variation and have limited power in capturing the enormous genetic diversity in Africans. Moreover, intrinsic differences in LD architecture between African and non-African populations restrict their ability to identify association signals in Africans.
To overcome these challenges the Human Heredity and Health in Africa (H3Africa) Consortium designed a 2.5 Million SNV-array as an Africa-centric, unbiased, cost-efficient solution for genomic research on the continent. An unprecedented dataset of over 2500 unpublished African whole genomes and novel computational approaches were used to design an array with a considerably better representation of African genetic diversity. Consequently, the H3Africa array has a dense coverage of African LD blocks and includes thousands of novel African variants. The array contains a comprehensive set of SNVs of potential clinical and pharmacogenomics significance based on databases (GWAS-Catalog, ClinVar, Cosmic and PharmGKB) and suggestions from local researchers.
Computational analyses show the H3Africa array to significantly outperform currently available arrays of similar size in terms of both genomic coverage and imputation accuracy. The array will provide researchers a resource to discover novel disease associations and also to narrow down on the causal variants in an African setting.
A Uganda innovation hub for deep analytics, disease surveillance, discovery science and translation
Presenter:
Beatrice Dhaala
MRC-Uganda
A Uganda innovation hub for deep analytics, disease surveillance, discovery science and translation (NULL) In recent years, health data has seen a dramatic increase in volume as well as complexity and diversity; mining this vast pool of complex health data has the potential among others to provide better insights into disease outbreak patterns, inform therapy target discovery and support health policy by improving our understanding of health and disease at a population level. The generation of data resources within the continent has created the need for initiatives supporting the expansion of expertise on health data management and analytics to ensure that Africa can fully benefit from health data. <br>The MRC/UVRI Uganda Medical Informatics centre (UMIC) was created as an initiative to achieve the broader development goals by harnessing the data and analytics revolution in Africa; in this context, the MRC/UVRI UMIC was set forth to capitalise on the potential benefits of large-scale health data by combining expertise on genomics, analytics, High Performance Compute (HPC) and DevOps with the routine use of high-end HPC resources capable of handling the current complexity and size of health care data in Africa. <br>Under this framework the UMIC supports major Pan-African and local research initiatives in medical genomics and bioinformatics such as The Genome Diversity in Africa Project (GDAP), TrypanoGEN (TPG), The PANGEA-HIV Consortium, The International AIDS Vaccine Initiative and The MRC/UVRI Uganda Research Unit on AIDS (MRC/UVRI). <br>As Global Health paradigms begin to shift, moving the focus from treatment to prevention, the UMIC intends to provide the tools to impact both foreign as well as domestic policy development.
- top -
ISCB Africa ASBCB Conference on Bioinformatics 2017
SESSION 2: Host/pathogen interactions and dynamics of infectious diseases
Oral Presentation Abstracts
The fecal microbiota of scavenging pigs (Sus scrofa domesticus) are potential reservoir of pathogens of public health importance
Presenter:
Kilaza Mwaikono
University of Cape Town
Additional authors:
Paul Gwakisa
Nelson Mandela African Institution of Sciences and Technology
School of Life Sciences and Bioengineering
Solomon Maina
International Livestock Research Institute
Biosciences eastern and central Africa
Pigs scavenging in dumpsites are exposed to microbes of public health importance, yet little is known of their fecal microbiota. We characterized the fecal microbiota of pigs scavenging in municipal dumpsite as compared with conventionally reared pigs.
Fifty-five fecal samples were collected from pigs under three management systems: Pigs scavenging on a dumpsite (FecD), indoor-reared (FecI) and pigs transferred from indoor to free-range on dumpsite (FecIF). Total DNA was extracted; V4 of 16S rRNA gene amplified and sequenced using MiSeq. The same sample was used to isolate enteric bacteria that were later tested to eight antibiotics.<br>A total of 4,364,507 sequences passed quality control from which 40,803 OTUs dominated by Firmicutes 51%, Proteobacteria 26%, Bacteroidetes 12%, Spirochaetes 3% and Actinobacteria 3% were detected. There was significant difference in bacterial community membership and structure between FecD and FecI (Yue and Clayton, P = 0.001; Jaccard, P = 0.032), respectively. Out of 830 genera, 74 were significantly different (P ≤ 0.05) between FecD and FecI amongst which 27 (36%) were exclusive to FecD. Some isolates from scavenging pigs had 99% sequence similarity to pathogenic Escherichia furgosonii, Shigella sonnei, Enterococcus faecium and Escherichia coli O154:H4. Over 50% of the isolates were resistant to Penicillin G, Ceftazidime and Nalidixic Acid, while others were multidrug resistant.
The fecal microbiota of pigs scavenging on dumpsite is a potential reservoir of pathogens. Controlled grazing of livestock to mitigate health risks is advocated. Further study of the gut microflora of free-range pigs and their clinical significance is warranted.
New Bioinformatics-based Discrimination Formulas for Differentiation of Thalassemia Traits from Iron Deficiency Anemia
Presenter:
Abdul Hafeez
Kandhro Mahidol University
Thalassemia traits and iron deficiency anemia (IDA) are the commonest disorders that frequently cause hypochromic microcytic anemia (HMA). Early screening of TTs and IDA is crucial for further investigation, therapeutic management, and genetic counseling. The present study aimed to differentiate TTs from IDA by analyzing discrimination formulas. The present study provides comprehensive data on hemoglobin disorders prevalent in five districts of Sindh Province, Pakistan. Among twelve published discrimination formulas, six formulas; MI, EF, G&K, RDWI, R, and HHI were the most reliable with higher accuracies with Youden’s index 1.0, and could discriminate TTs from IDA. The failure cutoff values of the six formulas were improved by the RF decision tree approach. Moreover, the S&L formula which completely failed to discriminate IDA from TTs with original cutoff value (<1530), improved with the use of new proposed cutoff value (<1016) and was found to successfully discriminate all cases of TTs from those with IDA. In addition, two newly proposed formulas discriminated TTs from IDA more reliably than the original 12 formulas assessed. Proposed formulas could play a crucial role for clinicians to discriminate between TTs and IDA, which both tend to have HMA, especially in premarital screening.
Protein Complexes of RNA-Seq gene expression data revealed functions enriched in the invasion of Red Blood Cells by Plasmodium falciparum
Presenter:
Jumoke Soyemi
Federal Polytechnic, Ilaro
Additional authors:
Itunuoluwa Isewon
Covenant University Bioinformatics Research Group
Computer and Information Sciences
Jelili Oyeladeuk
Covenant University,
Department of Computer and Information Sciences
Ezekiel Adebiyi
Covenant University
Department of Computer and Information Sciences
This study employed RNA-seq time course gene expression data specifically, at the late trophozoite and schizont stages of the Plasmodium falciparum where merozoites matures, ruptures and are released to invade the red blood cells. We extracted differentially expressed genes (DEGs) from a time series RNA-seq gene expression experiment using R package. Protein-protein interactions (PPIs) were identified from high-throughput yeast two-hybrid (Y2H) experiment and supplemented with PPIs from database. From the protein interaction network (PIN) built, we identified 16 protein complexes using the molecular complex detection (MCODE) algorithm in Cytoscape. The functional enrichment of the identified protein complexes revealed functions related to gene expression, translation, RNA transport and metabolic/biological processes. Identifying these protein interactions complexes are important because most proteins carry out their functions by interacting with other proteins since proteins rarely act alone. The identified protein complexes could further be taken to the laboratory as drug targets and validated in order to progress to the drug discovery phase for future therapy development against the disease. Quite a number of our predictions are consistent with those from previous studies. Protein complexes that are active during RBC invasion by the merozoites have been predicted by this study.
Keywords: RNA-Seq time course data, Plasmodium falciparum, DEGs, Protein Complexes, MCODE, Cytoscape
Endosymbiosis, origins and gene expression in the photosynthetic protist Euglena gracilis
Presenter:
ThankGod Ebenezer
University of Cambridge
Additional authors:
Martin Zoltner
University of Dundee
1Wellcome Trust Centre for Anti-Infectives Research
Alana Burrel
Oxford Brookes University
Department of Biological and Medical Sciences
Anna Nenarokova
Czech Academy of Sciences Biology Centre, Institute of Parasitology
Anna Vanclová
Charles University in Prague
Department of Parasitology
Binod Prasad
University of Erlangen-Nuremberg
Department of Biology
Carlos Santana
Universidad Pablo de Olavide
Centro Andaluz de Biología del Desarrollo (CABD)
Ellis O’Neill
University of Oxford
Department of Plant Sciences
Gowthaman Ramasamy
University of Washington
Center for Infectious Disease Research
Nerissa Nankissoor
University of Alberta
Department of Cell Biology
Samuel Obado
The Rockefeller University
Laboratory of Cellular and Structural Biology
Andrew Jackson
University of Liverpool
Department of Infection Biology, Institute of Infection and Global Health
Damien Devos
Universidad Pablo de Olavide
Centro Andaluz de Biología del Desarrollo (CABD)
Joel Dacks
University of Alberta
Department of Cell Biology
Julius Lukes
Czech Academy of Sciences
Biology Centre, Institute of Parasitology
Michael Lebert
University of Erlangen-Nuremberg
Department of Biology
Peter Myler
University of Washington
Center for Infectious Disease Research
Susan Vaughan
Oxford Brookes University
Department of Biological and Medical Sciences
Vladimir Hampl
Charles University in Prague
Department of Parasitology
Mark Carrington
University of Cambridge
Department of Biochemistry
Michael Ginger
University of Huddersfield
Department of Chemical and Biological Sciences
Steven Kelly
University of Oxford
Department of Plant Sciences
Mark Field
University of Dundee
Wellcome Trust Centre for Anti-Infectives Research
The photosynthetic flagellate Euglena gracilis harbours a secondary endosymbiotic plastid and is a distant relative of pathogenic trypanosomatids, a major component of global aquatic ecosystems and of considerable biotechnological potential with resistance to harsh conditions. Here we report genome, transcriptome and proteome drafts for E. gracilis. The genome is over 2Gb and has a coding potential of 36,526 predicted ORFs. Less than 25% of the genome is single copy sequence, indicating extensive repeat elements. Several gene families likely associated with the cell surface and signal transduction possess very large numbers of lineage-specific paralogs, suggesting great flexibility in environmental monitoring and, together with divergent mechanisms for metabolic control, novel solutions to adaptation to extreme environments. There are clear contributions from photosynthetic eukaryotes to the nuclear genome with red, green and brown algael genes evident, together with orthogroups shared with only trypanosomes and also with other excavates. Furthermore, we demonstrate that the majority of control of protein expression level is post-transcriptional despite the presence of conventional introns, that mRNA metabolism is highly unusual in transcriptional and nuclear export mechanisms and which differentiate Euglenids from the trypanosomatids. These data are a major advance in the understanding of the nuclear genome of Euglenids and provide a platform for investigation of the contributions of Euglena gracilis and relatives to the biosphere.
Protein Complex Detection from Plasmodium falciparum’s Interactome using MCODE Algorithm
Presenter:
Trust Odia
Covenant University Bioinformatics Research Group
Additional authors:
Jelili Oyelade
Covenant University Bioinformatics Research Group
Computer and Information Sciences
Itunuoluwa Isewon
Covenant University Bioinformatics Research Group Computer and Information Science
Solomon Rotimi
Covenant University Bioinformatics Research Group
Biological Sciences
Ezekiel Adebiyi
Covenant University Bioinformatics Research Group Computer and Information Sciences
From high-throughput experimental analysis, more than 50% of the proteins in Plasmodium falciparum interactome have been identified with unknown functions. The comprehensive description of the parasite’s protein interaction network revealed that 82% of the interactions included at least one protein annotated as unknown, 33% of the interactions included two unknown proteins.
In this paper, we used a graph-based clustering algorithm for detecting molecular complexes in the parasite’s interactome. For functional annotation, we performed sequence similarity alignment, protein structure alignment, phylogenetic analysis and gene functional enrichment to predict function for identified unknown proteins and we were able to predict functions for PFL1395c and PFL0350c proteins. PFL1395c is predicted to be a histone acetyltransferease enzyme that regulates gene expression in Plasmodium falciparum, while PFL0350c is predicted to be carrying out DNA binding and bending of linear DNA distorted structures that facilitate binding of nucleoprotein complexes. We used MCODE algorithm to get insight into the functions of two previously uncharacterized proteins, which is relevant in understanding the molecular mechanism of the parasite. Understanding protein functions plays a great role in protein structure prediction, molecular biology, molecular mechanism of cellular processes and drug design.
Leveraging viral phylodynamics to inform spatiotemporal transmission of viral infectious disease in Africa: 2009 Influenza A/H1N1 in Africa
Presenter:
Fredrick Nindo
University of Cape Town
Background
Emerging and re-emerging viral pathogens pose serious health threats to both humans and livestock. Although all global regions are at risk of viral infectious disease outbreaks, the African continent is disproportionately disadvantaged due to its weak health systems to mitigate, manage and prevent further spread of the pathogens in an outbreak situation.
Molecular sequence data in combination with ecological, economic and demographic factors have proven to robustly describe phylodynamics of viral infectious diseases.
Human mobility plays a central role in infectious disease transmission and due to economic constraints probably road transport networks are the key drivers of infectious disease spread in Africa rather than air travel, railway networks or Euclidean distances between sampling locations
Aims and Objectives
This study sought to unravel the introduction, the spatial dispersal pattern and to evaluate the contribution of genetic, ecological, and economic predictors of viral infectious transmission patterns using the case of 2009 Influenza A/H1N1 pandemic virus on the African continent.
Materials and Methods
In this study, we combined ecological, economic and demographic data Bayesian phylogenetics to unravel the introduction, dispersal and test predictors of viral infectious diseases transmission patterns using the case of 2009 Influenza A/H1N1 pandemic virus on the African continent.
Results, Discussion and Conclusions
Our study suggested that there were multiple simultaneous introductions of the 2009 H1N1 pandemic virus into the African continent and the phylogenetically inferred probable source populations existed in North America and Asia. Transmission predictor analysis suggested that proximity (geographical distance), air travel, and sampling location latitude might have contributed significantly to the spread of the 2009 Influenza A/H1N1pandemic virus
Endosymbiosis, origins and gene expression in the photosynthetic protist Euglena gracilis
Presenter:
ThankGod Ebenezer
University of Cambridge
Additional authors:
Martin Zoltner
University of Dundee
1Wellcome Trust Centre for Anti-Infectives Research
Alana Burrel
Oxford Brookes University
Department of Biological and Medical Sciences
Anna Nenarokova
Czech Academy of Sciences Biology Centre, Institute of Parasitology
Anna Vanclová
Charles University in Prague
Department of Parasitology cz::
Binod Prasad
University of Erlangen-Nuremberg
Department of Biology
Carlos Santana
Universidad Pablo de Olavide
Centro Andaluz de Biología del Desarrollo (CABD)
Ellis O’Neill
University of Oxford
Department of Plant Sciences
Gowthaman Ramasamy
University of Washington
Center for Infectious Disease Research
Nerissa Nankissoor
University of Alberta
Department of Cell Biology
Samuel Obado
The Rockefeller University
Laboratory of Cellular and Structural Biology
Andrew Jackson
University of Liverpool
Department of Infection Biology, Institute of Infection and Global Health
Damien Devos
Universidad Pablo de Olavide
Centro Andaluz de Biología del Desarrollo (CABD)
Joel Dacks
University of Alberta
Department of Cell Biology
Julius Lukes
Czech Academy of Sciences
Biology Centre, Institute of Parasitology
Michael Lebert
University of Erlangen-Nuremberg
Department of Biology
Peter Myler
University of Washington
Center for Infectious Disease Research
Susan Vaughan
Oxford Brookes University
Department of Biological and Medical Sciences
Vladimir Hampl
Charles University in Prague
Department of Parasitology
Mark Carrington
University of Cambridge
Department of Biochemistry
Michael Ginger
University of Huddersfield
Department of Chemical and Biological Sciences
Steven Kelly
University of Oxford
Department of Plant Sciences
Mark Field
University of Dundee
Wellcome Trust Centre for Anti-Infectives Research
The photosynthetic flagellate Euglena gracilis harbours a secondary endosymbiotic plastid and is a distant relative of pathogenic trypanosomatids, a major component of global aquatic ecosystems and of considerable biotechnological potential with resistance to harsh conditions. Here we report genome, transcriptome and proteome drafts for E. gracilis. The genome is over 2Gb and has a coding potential of 36,526 predicted ORFs. Less than 25% of the genome is single copy sequence, indicating extensive repeat elements. Several gene families likely associated with the cell surface and signal transduction possess very large numbers of lineage-specific paralogs, suggesting great flexibility in environmental monitoring and, together with divergent mechanisms for metabolic control, novel solutions to adaptation to extreme environments. There are clear contributions from photosynthetic eukaryotes to the nuclear genome with red, green and brown algael genes evident, together with orthogroups shared with only trypanosomes and also with other excavates. Furthermore, we demonstrate that the majority of control of protein expression level is post-transcriptional despite the presence of conventional introns, that mRNA metabolism is highly unusual in transcriptional and nuclear export mechanisms and which differentiate Euglenids from the trypanosomatids. These data are a major advance in the understanding of the nuclear genome of Euglenids and provide a platform for investigation of the contributions of Euglena gracilis and relatives to the biosphere.
- top -
ISCB Africa ASBCB Conference on Bioinformatics 2017
SESSION 1: H3ABioNet highlights
Oral Presentation Abstracts
Variants calling optimization: parameter sweep of the GATK best practices pipeline
Presenter:
Azza Ahmed
University of Khartoum
Additional authors:
Gloria Rendon
University of Illinois Urbana-Champaign
Institute for Genomic Biology
Liudmila Sergeevna Mainzer
University of Illinois Urbana-Champaign
Institute for Genomic Biology, National Center for Supercomputing Applications
Victor Jongeneel
University of Illinois Urbana-Champaign
Institute for Genomic Biology
Faisal M. Fadlelmola
University of Khartoum
Centre for Bioinformatics and Systems Biology
The advent of massively parallel sequencing technologies (Next Generation Sequencing, NGS) had modified the landscape of human genetics. Whole Exome Sequencing (WES) is the NGS branch that focuses on the exonic regions of the eukaryotic genomes. Exomes are of interest as they are helping us understand high-penetrance allelic variation and its relationship to phenotype.
Variant calling is a study of genomic sequences differences, between some samples of interest and the reference genome; with the purpose of aiding understanding disease (or phenotype mechanism) and ultimately designing optimal treatment targets (i.e. personalized medicine). Typically, this involves many wet lab assays and procedures for preparing the biological samples and intensive computational processing via many tools and software. Errors can creep into the analysis from any of these aspects.
When carried out in large cohorts, errors are exacerbated, and the variants observed at the level of the individual are lost in joint genotyping. Besides experimental wet lab errors, the called variants are subject to biases due to the choice of software, configuration of the analysis pipeline, and individual parameters of each tool used. Also intended as a pilot phase of a collaboration with Mayo Clinic in Florida, USA, this talk provides insights into the effect of the parameter configurations in a variant calling pipeline following GATK best practices from a mathematical point of view, along with experimental results, with the objective of identifying optimization targets in such a set up. Computational challenges relating to running the pipeline in this context are also highlighted.
Comparative analysis of existing pipelines for assessment of arbuscular mycorrhizal fungal biodiversity in natural and commercial Rooibos (Aspalathus linearis) and Honeybush (Cyclopia intermedia) soil samples
Presenter:
Herna de Wit
Rhodes University
A mutually beneficial association exists between the fungal phylum Glomeromycota and higher plant roots. Arbuscular mycorrhizal (AM) fungi are important in performing various ecological functions in exchange for host photosynthetic carbon contributing to host plant’s fitness. AM fungi are unculturable and although their many benefits have been well documented, their biodiversity is not studied in detail.
Two types of plant hosts were studied: Rooibos (Aspalathus linearis) and Honeybush (Cyclopia intermedia). Both are popular teas in South Africa and have a growing worldwide market due to their medicinal properties. Ribosomal sequences (18S), were generated using 454 pyrosequencing at Rhodes University. Soil samples were collected from both natural and commercial plant populations. The sequence data required rationalization with the identification of a suitable pipeline in order to determine changes in AM fungal biodiversity.
As AM fungi are sensitive to many agricultural practices, a higher diversity and abundance of AM fungi were expected in soils from natural rather than in commercial soil samples. None of the bioinformatics pipelines were able to provide suitable AM fungal biodiversity data, due to lack of available databases. A suitable pipeline, to effectively analyse 18S AM fungal data needs to be designed in order to further investigate changes in biodiversity of these important soil fungi.
A Metagenomic Approach to the Characterization of the Microbiomes of Sickle Cell Disease-associated Leg Ulcers
Presenter:
Deborah Fasesan
National Biotechnology Development Agency
Additional authors:
Jessica Holmes
University of Illinois at Urbana-Champaign
High Performance Biological Computing Group
Jenny Zadeh
University of Illinois at Urbana-Champaign
High Performance Biological Computing Group
Ayodele Ogunkeyede
University of Ilorin
Department of Surgery
Gladys Falusi
University of Ibadan
Institute for Advanced Medical Research and Training
Victor Jongeneel
University of Illinois at Urbana-Champaign
High Performance Biological Computing Group
Oyekanmi Nash
National Biotechnology Development Agency
Centre for Genomics Research and Innovation
Scientific improvements in microbial evaluation have led to more accurate analysis of microbiomes. Such improvements include metagenomics- a field which explores microorganisms in their natural habitat without the need for conventional plate cultivation. Microbiome studies involve the use of 16S rRNA analysis for bacterial composition and can also include examination of ITS regions for the determination of fungal community within an environment.<br>In this study, the microbiomes of leg ulcers of sickle cell disease subjects were determined in the outcome of compression therapy treatment (CT). DNA from wound samples were extracted, amplified using appropriate bacterial and fungal primers for selected regions (V3-V5 and V4 segments of the 16s rRNA bacterial region and ITS 3-4 region of the fungal genomes) and libraries were constructed for paired end sequencing. Generated sequences were then subjected to microbiome analysis using the metagenomic pipeline QIIME (Quantitative Insights Into Microbial Ecology).
Bacterial taxonomic classification indicated a decline in the occurrence of some pathogenic genera after four weeks of CT. Although, Stenotrophomonas, Bradyrhizobium and Burkholderia were seen in samples collected after treatment, results showed significant reduction in wound sizes after treatment. Fungal taxonomic classification revealed the dominance of Aspergillus, Candida and Wickerhamomyces but with the occurrence of plant like material seen in samples before treatment. This study highlights the possible positive contribution of CT on sickle cell leg ulcers. The association between these microorganisms, treatment method applied, as well as the effect of the combination of antibiotics and CT on sickle cell leg ulcers needs further determination.
A Uganda innovation hub for deep analytics, disease surveillance, discovery science and translation . In recent years, health data has seen a dramatic increase in volume as well as complexity and diversity; mining this vast pool of complex health data has the potential among others to provide better insights into disease outbreak patterns, inform therapy target discovery and support health policy by improving our understanding of health and disease at a population level. The generation of data resources within the continent has created the need for initiatives supporting the expansion of expertise on health data management and analytics to ensure that Africa can fully benefit from health data.
The MRC/UVRI Uganda Medical Informatics centre (UMIC) was created as an initiative to achieve the broader development goals by harnessing the data and analytics revolution in Africa; in this context, the MRC/UVRI UMIC was set forth to capitalise on the potential benefits of large-scale health data by combining expertise on genomics, analytics, High Performance Compute (HPC) and DevOps with the routine use of high-end HPC resources capable of handling the current complexity and size of health care data in Africa.
Under this framework the UMIC supports major Pan-African and local research initiatives in medical genomics and bioinformatics such as The Genome Diversity in Africa Project (GDAP), TrypanoGEN (TPG), The PANGEA-HIV Consortium, The International AIDS Vaccine Initiative and The MRC/UVRI Uganda Research Unit on AIDS (MRC/UVRI).
As Global Health paradigms begin to shift, moving the focus from treatment to prevention, the UMIC intends to provide the tools to impact both foreign as well as domestic policy development.
Comparative analysis of existing pipelines for assessment of arbuscular mycorrhizal fungal biodiversity in natural and commercial Rooibos (Aspalathus linearis) and Honeybush (Cyclopia intermedia) soil samples.
A mutually beneficial association exists between the fungal phylum Glomeromycota and higher plant roots. Arbuscular mycorrhizal (AM) fungi are important in performing various ecological functions in exchange for host photosynthetic carbon contributing to host plant’s fitness. AM fungi are unculturable and although their many benefits have been well documented, their biodiversity is not studied in detail.<br><br>Two types of plant hosts were studied: Rooibos (Aspalathus linearis) and Honeybush (Cyclopia intermedia). Both are popular teas in South Africa and have a growing worldwide market due to their medicinal properties. Ribosomal sequences (18S), were generated using 454 pyrosequencing at Rhodes University. Soil samples were collected from both natural and commercial plant populations. The sequence data required rationalization with the identification of a suitable pipeline in order to determine changes in AM fungal biodiversity.
As AM fungi are sensitive to many agricultural practices, a higher diversity and abundance of AM fungi were expected in soils from natural rather than in commercial soil samples. None of the bioinformatics pipelines were able to provide suitable AM fungal biodiversity data, due to lack of available databases. A suitable pipeline, to effectively analyse 18S AM fungal data needs to be designed in order to further investigate changes in biodiversity of these important soil fungi.
Integrative analysis of cervical cancer multi-omics data: Towards novel biomarkers discovery
Presenter:
Somia Mohammed
Centre for Bioinformatics & Systems Biology
Additional authors:
Faisal Fadlelmola
Centre for Bioinformatics & Systems Biology (CBSB)
Alia Benkahla
Institute Pasteur of Tunis
bioMathematics and bioStatistics (BIMS)
Mohssin Abdalla
Faculty of Mathematical Sciences
Computer Science
Cervical cancer is the second most common type of cancer in women worldwide. Due to poor access to screening and treatment services, more than 90% of deaths occur in women living in low- and middle- income countries. Although a diagnostic tool (PapTest) is widely available, cervical cancer incidence still remains high worldwide, and especially in developing countries, attributed to a large extent to sensitivities of the Pap test and unavailability of test in developing countries.
In this study, multi-omics data analysis was evaluated by integrating array-CGH, SNP array and gene expression data in cervical cancer. A meta-analysis approach was adopted whereby cervical cancer multi-omics datasets from Gene Expression Omnibus (GEO), aCGH, gene expression and SNP array, were selected to set up the analysis. Each data set was analyzed separately by implementing R. The dataset were subject to statistical tests (e.g. ANOVA and T-test).
Chromosomal regions showed significant amplification in most of the samples in chromosomes 1,3 and 19 whereas losses were found in 11, 4 and 13. The aCGH data is searched for genes with a known regulatory role whose copy number is altered in the samples. The matched transcriptomics data is then examined to see if a gene's altered copy number is associated with a concurrent change in the gene's expression, thus adding weight to the argument that the gene may be contributing to cervical cancer. The list of genes that show strong aCGH/expression is subjected to various public databases (e.g. OMIM) to perform further in silico analysis and validation.
DREAM of Malaria: an open ecosystem to predict drug sensitivity in malaria parasites
Presenter:
Amel Ghouila
Institut Pasteur de Tunis
Additional authors:
Katrina Button-Simons
University of Notre Dame
Eck Institute for Global Health
Jean-Baka Domelevo Entfellner
University of the Western Cape
South African National Bioinformatics Institute
Sumir Panji
University of Cape Town
Computational Biology Division, Department of Integrative Biomedical Sciences, Institute of Infectious Disease and Molecular Medicine
Geoffrey Siwo
IBM Research Africa
Johannesburg Lab
Sage Davis
University of Notre Dame
Eck Institute for Global Health
Faisal Fadlelmola
University of Khartoum
Centre for Bioinformatics and Systems Biology, Faculty of Science
The DREAM of Malaria Hackathon Participants:
Michael Ferdig
University of Notre Dame
Eck Institute for Global Health
Nicola Mulder
University of Cape Town
Computational Biology Division
Department of Integrative Biomedical Sciences
Institute of Infectious Disease and Molecular Medicine
Collaborative analysis challenges such as DREAM have brought considerable computational expertise to cancer and autoimmune disease research, showing that aggregating results across methods outperforms single methods. Unfortunately, even interdisciplinary collaborations often lack the resources necessary to employ multiple complex analyses. To encourage individual labs to create datasets for challenges; H3ABioNet, IBM Africa and the Ferdig lab (University of Notre Dame) teamed up to conduct a hackathon to explore whether the transcriptomic profile can be used to predict drug sensitivity in malaria parasites. The hackathon had two goals: prepare data for analysis and detect the presence of sufficient signal in the dataset to predict drug susceptibility.
Twenty-three researchers and Ph.D. students from across Africa participated in the hackathon in September, 2016. Three teams were formed with complementary areas of expertise and diverse skills. Initial exploration of the data identified unexpected issues. One team identified eight outliers originating from the same microarray which were then re-hybridized correcting the issue. Another team demonstrated that genome-wide differences existed between and among isolates and that intra-erythrocytic developmental cycle (IDC) staging was a significant source of biological variation.
Teams designed regression models (ensemble partial least squares, regularized regression, random forests, etc) to predict drug sensitivity from gene expression features and covariates. Some predictive models gave promising results (e.g. training MSE = 0.072 and test MSE = 0.21, with 32 features selected). Overall, these results demonstrate it is challenging, but possible to extract signal from this dataset to predict the drug sensitivity of malaria isolates.
Designing a course model for distance-based online bioinformatics training in Africa: the H3ABioNet experience
Presenter:
Shaun Aron
University of the Witwatersrand
Sydney Brenner Institute for Molecular Bioscience
Additional authors:
Kim Gurwitz
University of Cape Town
Computational Biology Division
Sumir Panji
University of Cape Town
Computational Biology Division
Suresh Maslamoney
University of Cape Town
Computational Biology Division
Pedro L. Fernandes
Instituto Gulbenkian de Ciência Bioinformatics pt::
David P. Judge
Independent Bioinformatics Training Specialist
Bioinformatics uk::
Amel Ghouila
Institut Pasteur de Tunis Laboratory Transmission, Control and Immunobiology of Infections tn::
Jean-Baka Domelevo Entfellner
University of the Western Cape South African National Bioinformatics Institute/MRC Unit for Bioinformatics Capacity Development
Fatma Z. Guerfali
Institut Pasteur de Tunis Laboratory Transmission, Control and Immunobiology of Infections
Colleen Saunders
University of the Western Cape South African National Bioinformatics Institute/MRC Unit for Bioinformatics Capacity Development
Ahmed Mansour
Zagazig University
Genetics Department
Samson P. Salifu
Kwame Nkrumah University of Science and Technology
Department of Biochemistry and Biotechnology/Kumasi Centre for Collaborative Research
Rehab Ahmed
University of Khartoum/Future University of Suda
Centre for Bioinformatics and Systems Biology
Ruben Cloete
University of the Western Cape
South African National Bioinformatics Institute/MRC Unit for Bioinformatics Capacity Development
Jonathan Kayondo
Uganda Virus Research Institute Entomology
A distance-based Introduction to Bioinformatics course is run by H3ABioNet - a pan African Bioinformatics network for H3Africa. In its second iteration this year, the free-of-charge, 3-month, skills-based course teaches the basics of various bioinformatics analyses. It makes use of multiple education delivery methods, namely: face-to-face learning; distance learning; and open educational resources, in order to increase access across Africa. Local classrooms - 27 classrooms hosting roughly 600 participants, in total, across 12 African countries (2017) - are attended face-to-face for additional support where there is interaction with volunteer, teaching assistants and with peers. During these face-to-face sessions, classrooms watch open access, pre-recorded and downloaded lecture recordings, prepared by expert African bioinformatics trainers. Classrooms also sign in to a virtual classroom that links them all to each other and to the trainer during biweekly contact sessions. Further, course participants and volunteer local staff engage via online forums hosted on the course management platform. Additional features of the course this year, developed out of an extensive review of last year’s iteration, include: staff training at local classrooms; promoting previous year attendees as trainee teaching assistants; consolidation sessions; and encouraging engagement within and across classrooms as well as with local bioinformatics communities.
- top -
ISCB Africa ASBCB Conference on Bioinformatics 2017
PROMOTION
Click here to download .pdf

ISCB Africa ASBCB Conference on Bioinformatics 2017
COMMITTEE
Conference Chairs:
Alan Christoffels, University of the Western Cape, South Africa
Nicola Mulder, University of Cape Town, South Africa
Sumir Panji, H3ABioNet / Computational Biology, University of Cape Town, South Africa
Scientific Program Chairs:
Ruben Cloete, South African National Bioinformatics Institute, Cape Town, South Africa
Segun Fatumo, Wellcome Trust Sanger Institute Cambridge, UK
Amel Ghouila , Institute Pasteur of Tunis, Tunisia
Local Organizing Chair:
Jonathan Kayondo, Uganda Virus Research Institute, Uganda
Moses Kizza, Administrator, MUII-Plus/Uganda Virus Research Institute, Uganda
Allen Mukhwana, Centre Manager MUII-Plus/Uganda Virus Research Institute, Uganda
Conference Steering Committee:
Marion Adebiyi, Covenant University, Nigeria
Alia Benkahla, Institute Pasteur of Tunis, Tunisia
Alan Christoffels, University of the Western Cape, South Africa
Ruben Cloete, South African National Bioinformatics Institute, Cape Town, South Africa
Segun Fatumo, Wellcome Trust Sanger Institute Cambridge, UK
Amel Ghouila , Institute Pasteur of Tunis, Tunisia
Jonathan Kayondo, Uganda Virus Research Institute, Uganda
Janet Kelso MPI for Evolutionary Anthropology, Germany
Diane Kovats, ISCB, USA
Nicola Mulder, University of Cape Town, South Africa
Sumir Panji, H3ABioNet / Computational Biology, University of Cape Town
Burkhard Rost, TUM Munich, Germany
Natasha Wood, University of Cape Town, South Africa
- top -