ACCEPTED PAPERS
Updated Oct 28, 2014
The following papers will be presented as talks during the conference.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
A validated gene regulatory network and GWAS to identify early transcription factors in T-cell associated diseases
Mika Gustafsson1, Danuta Gawel1, Sandra Hellberg1, Aelita Konstantinell1, Daniel Eklund1, Jan Ernerudh1, Antonio Lentini1, Robert Liljenström1, Johan Mellergård1, Hui Wang2, Colm E. Nestor1, Huan Zhang1 and Mikael Benson1
1Linköpings Univeristet, 2MD Anderson Cancer Center
The identification of early regulators of disease is important for understanding disease mechanisms, as well as finding candidates for early diagnosis and treatment. Such regulators are difficult to identify because patients generally present when they are symptomatic, after early disease processes. Here, we present an analytical strategy to systematically identify early regulators by combining gene regulatory networks (GRNs) with GWAS. We hypothesized that early regulators of T-cell associated diseases could be found by defining upstream transcription factors (TFs) in T-cell differentiation. Time-series expression profiling identified upstream TFs of T-cell differentiation into Th1/Th2 subsets enriched for disease associated SNPs identified by GWAS. We constructed a Th1/Th2 GRN based on integration of expression, DNA methylation profiling and sequence-based predictions data using LASSO algorithm. The GRN was validated by ChIP-seq and siRNA knockdowns. GATA3, MAF and MYB were prioritized based on GWAS and the number of GRN predicted targets. The disease relevance was supported by differential expression of the TFs and their targets in profiling data from six T-cell associated diseases. We tested if the three TFs or their splice variants changed early in disease by exon profiling of two relapsing diseases, namely multiple sclerosis and seasonal allergic rhinitis. This showed differential expression of splice variants of the TFs during relapse-free asymptomatic stages. Potential targets of the splice variants were validated based on expression profiling and siRNA knockdowns. Those targets changed during symptomatic stages. Our results show that combining construction of GRNs with GWAS can be used to infer early regulators of disease.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Are all genetic variants in DNase I sensitivity regions functional?
Gregory A. Moyerbrailean1, Chris T. Harvey1, Cynthia A. Kalita1, Xiaoquan Wen2, Francesca Luca1, Roger Pique-Regi1
1Wayne State University, 2University of Michigan
A detailed mechanistic understanding of the direct functional consequences of DNA variation on gene regulatory mechanism is critical for a complete understanding of complex trait genetics and evolution. Here, we present a novel approach that integrates sequence information and DNase I footprinting data to predict the impact of a sequence change on transcription factor binding. Applying this approach to 653 DNase-seq samples, we identified 3,831,862 regulatory variants predicted to affect active regulatory elements for a panel of 1,372 transcription factor motifs. Using QuASAR, we validated the non-coding variants predicted to be functional by examining allele-specific binding (ASB). Combining the predictive model and the ASB signal, we identified 3,217 binding variants within footprints that are significantly imbalanced (20% FDR). Even though most variants in DNase I hypersensitive regions may not be functional, we estimate that 56% of our annotated functional variants show actual evidence of ASB. To assess the effect these variants may have on complex phenotypes, we examined their association with complex traits using GWAS and observed that ASB-SNPs are enriched 1.22-fold for complex traits variants. Furthermore, we show that integrating footprint annotations into GWAS meta-study results improves identification of likely causal SNPs and provides a putative mechanism by which the phenotype is affected.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
A scalable method for molecular network reconstruction identifies properties of targets and mutations in acute myeloid leukemia
Edison Ong1, Anthony Szedlak2, Yunyi Kang, Peyton Smith1, Nicholas Smith1, Madison McBride3, Darren Finlay3, Kristiina Vuori3, James Mason4, Edward D. Ball5, Carlo Piermarocchi2, Giovanni Paternostro3
1Salgomed, 2Michigan State University, 3Sanford-Burnham Medical Research Institute, 4Scripps Health, San Diego, 5University of California, San Diego
A key aim of systems biology is the reconstruction of molecular networks. However, we do not yet have networks that integrate information from all datasets available for a particular clinical condition. This is in part due to the limited scalability, in terms of required computational time and power, of existing algorithms. Network reconstruction methods should also be scalable in the sense of allowing scientists from different backgrounds to efficiently integrate additional data.
We present a network model of acute myeloid leukemia (AML). In the current version (AML 2.1) we have used gene expression data (both microarray and RNA-seq) from five different studies comprising a total of 771 AML samples and a protein-protein interactions dataset. Our scalable network reconstruction method is in part based on the well-known property of gene expression correlation among interacting molecules. The difficulty of distinguishing between direct and indirect interactions is addressed by optimizing the coefficient of variation of gene expression, using a validated gold standard dataset of direct interactions. Computational time is much reduced compared to other network reconstruction methods. A key feature is the study of the reproducibility of interactions found in independent clinical datasets.
An analysis of the most significant clusters, and of the network properties (intraset efficiency, degree, betweenness centrality, and PageRank) of common AML mutations demonstrated the biological significance of the network. A statistical analysis of the response of blast cells from eleven AML patients to a library of kinase inhibitors provided an experimental validation of the network. A combination of network and experimental data identified CDK1, CDK2, CDK4, CDK6, and other kinases as potential therapeutic targets in AML.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
A cell lineage-specific regulatory network inferred using limited expression data of erythropoiesis
Fan Zhu1, Lihong Shi1, James Engel1, Yuanfang Guan1
1University of Michigan
Modeling regulatory networks using expression data observed in a differentiation process may help identify context-specific interactions. Despite intensive research efforts on this topic, the outcome of the current algorithms highly depends on the quality and quantity of a single time-course data, and the performance may be compromised for data with a limited number of samples. In this work, we report a novel multi-layer graphical model that is capable of leveraging heterogeneous, generic, publicly available time-course datasets, as well as limited cell lineage-specific data to model regulatory networks specific to a differentiation process. First, a collection of network inference methods are used to predict the regulatory relationships in individual datasets. Then, the inferred relationships are weighted and integrated together by evaluating against the cell lineage-specific data. To test the accuracy of this algorithm, we collected a time-course RNA-Seq dataset during human erythropoiesis to infer regulatory relationships specific to this differentiation process. The resulting erythroid-specific regulatory network reveals novel regulatory relationships activated in erythropoiesis, which were further validated by genome-wide TR4 binding studies using ChIP-seq. These erythropoiesis-specific regulatory relationships were not identifiable by single dataset-based methods or context-independent integrations. Analysis of the predicted targets reveals that they are all closely associated with hematopoietic lineage differentiation. In summary, this paper develops an integrative strategy that is capable of leveraging a limited, cell type-specific expression dataset and large-scale, generic time-course datasets to infer regulatory networks specific to a differentiation process, which is applicable to other cell lineages.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
cDREM: inferring dynamic combinatorial gene regulation
Aaron Wise1, Ziv Bar-Joseph1
1Carnegie Mellon University
Motivation: Genes are often combinatorially regulated by multiple transcription factors (TFs). Such combinatorial regulation plays an important role in development and facilitates the ability of cells to respond to different stresses. While a number of approaches have utilized sequence and ChIP based datasets to study combinational regulation, these have often ignored the combinational logic and the dynamics associated with such regulation.
Results: Here we present cDREM, a new method for reconstructing dynamic models of combinatorial regulation. cDREM integrates time series gene expression data with (static) protein interaction data. The method is based on a hidden Markov model and utilizes the sparse group Lasso to identify small subsets of combinatorially active TFs, their time of activation and the logical function they implement. We tested cDREM on yeast and human data sets. Using yeast we show that the predicted combinatorial sets agree with other high throughput genomic datasets and improve upon prior methods developed to infer combinatorial regulation. Applying cDREM to study human response to flu we were able to identify several combinatorial TF sets, some of which were known to regulate immune response while others represent novel combinations of important TFs.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Multi-species network inference improves gene regulatory network reconstruction for early embryonic development in Drosophila
Anagha Joshi1, Yvonne Beck1, Tom Michoel1
1The Roslin Institute, University of Edinburgh
Gene regulatory network inference uses genome-wide transcriptome measurements in response to genetic, environmental or dynamic perturbations to predict causal regulatory influences between genes. We hypothesized that evolution also acts as a suitable network perturbation and that integration of data from multiple closely related species can lead to improved reconstruction of gene regulatory networks. To test this hypothesis, we predicted networks from temporal gene expression data for 3,610 genes measured during early embryonic development in six Drosophila species, and compared predicted networks to gold standard networks of ChIP-chip and ChIP-seq interactions for developmental transcription factors in five species. We found that (i) the performance of single-species networks was independent of the species where the gold standard was measured; (ii) differences between predicted networks reflected the known phylogeny and differences in biology between the species; (iii) an integrative consensus network which minimized the total number of edge gains and losses with respect to all single-species networks performed better than any individual network. Our results show that in an evolutionarily conserved system, integration of data from comparable experiments in multiple species improves the inference of gene regulatory networks. They provide a basis for future studies on the numerous multi-species gene expression datasets for other biological processes available in the literature.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Reconstruction of gene regulatory networks based on repairing sparse low-rank matrices
Young Hwan Chang1, Roel Dobbe1, Palak Bhushan1, Joe W. Gray2, Claire J. Tomlin1
1University of California, Berkeley, 2Oregon Health and Science University
With the growth of high-throughput proteomic data, in particular time series gene expression data from various perturbations, a general question that has arisen is how to organize inherently heterogenous data into meaningful structures. Since biological systems such as breast cancer tumors respond differently to various treatments, little is known about exactly how these gene regulatory networks (GRNs) operate under different stimuli. For example, when we apply a drug-induced perturbation to a target protein, we often only know that the dynamic response of the specific protein may be affected. We do not know by how much, how long and even whether this perturbation affects other proteins or not. Challenges due to the lack of such knowledge not only occur in modeling the dynamics of a GRN but also cause bias or uncertainties in identifying parameters or inferring the GRN structure. This paper describes a new algorithm which enables us to estimate bias error due to the effect of perturbations and correctly identify the common graph structure among biased inferred graph structures. To do this, we retrieve common dynamics of GRN subject to various perturbations. We refer to the task as “repairing” inspired by “image repairing” in computer vision. The method can automatically correctly repair the common graph structure across perturbed GRNs, even without precise information about the effect of the perturbations. We evaluate the method on synthetic data sets and demonstrate advantages over l1-regularized graph inference by advancing our understanding of how these networks respond across different targeted therapies.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Pathways on demand: automated reconstruction of human signaling networks
Anna Ritz1, Christopher Poirel1, Allison Tegge1, Nicholas Sharp1, Allison Powell1, Kelsey Simmons1, Shiv Kale1, T.M. Murali1
1Virginia Polytechnic Institute and State University
Signaling pathways are a cornerstone of systems biology. Several databases store representations of these pathways that are amenable for automated analyses. Despite painstaking manual curation, significant variations exist between databases. To overcome these limitations, we present PathLinker, a new computational method that can reconstruct a signaling pathway from a background protein interaction network given only the identities of the receptors and transcription factors and regulators in that pathway. We demonstrate that PathLinker can reconstruct the Wnt pathway in the NetPath database with much higher precision and recall than several state-of-the-art algorithms, recovering non-canonical branches that appear only in this pathway's representation in other databases. PathLinker suggests a surprising role for CFTR, a chloride ion channel transporter of the ABC class, in Wnt/beta-catenin signaling, which we validate using siRNA experiments. We extend our computational results to accurately reconstruct a comprehensive set of signaling pathways in the NetPath database. We demonstrate that PathLinker can bridge differing representations of the same pathway between databases.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Inferring the genome-wide functional modulatory network: a case study on the NF-κB/RelA transcription factor
Xueling Li1, Min Zhu2, Allan Brasier1, Andrzej Kudlicki1
1University of Texas Medical Branch at Galveston, 2Hefei Institutes of Physical Science, Chinese Academy of Sciences
How different pathways lead to the activation of a specific transcription factor with specific effects is not fully understood. A modulatory network is composed of triplets of a specific transcription factor, target genes and modulators. Modulators usually affect the activity of the specific transcription factor at the post-transcription level in a target gene-specific manner (action mode), which may be classified as enhancement, attenuation and inversion of the activation or inhibition. Reconstructing such modulatory network will help to interpret how transcription factors produce distinct gene responses to different stimuli. As a case study, here we inferred, from a large collection of expression profiles, all potential modulations of NF-κB/RelA. The predicted modulators include many proteins previously not reported as physically binding to RelA. The functions of the predicted modulators are consistent with biological activities of NF-κB/RelA include RNA processing, alternative splicing, cell cycle, mitochondrion, ubiquitin-dependent proteolysis and ribosome biogenesis, and are consistent with binding modulators in our previous study. The predicted genome-wide RelA modulators from different enriched pathways or processes exert specific prevalent action modes on distinct pathways through RelA. Also, the modulators from non coding RNA (ncRNA), RNA binding proteins, transcription factors, cytoskeleton, and kinases modulate the NF-κB/RelA activity with specific action modes consistent with their molecular functions and modulation level. Finally, we analyzed the modulatory network of NF-κB/RelA in the context of TGFB1 induced epithelial-mesenchymal transition (EMT). Here modulators of NF-κB/RelA included those involved in extracellular matrix (FBN1), cytoskeletal regulation (ACTN1) and tumor suppression (FOXP1).
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Systematic study of synthetic transcript features in S. cerevisiae exposes gene-expression determinants
Tuval Ben-Yehezkel1, Shimshi Atar2, Tzipy Marx1, Rafael Cohen1, Alon Diament2, Alexandra Dana2, Anna Feldman2, Ehud Shapiro1, Tamir Tuller2
1Weizmann Institute of Science, 2Tel Aviv University
A major challenge in functional genomics is understanding how different parts of the transcript affect aspects of its expression. Heterologous gene expression can potentially contribute to this research topic, but has rarely been studied systematically, specifically in eukaryotes. Here, we use a synthetic biology approach to study the distinct and causal effect of different parts of the transcript in the eukaryote S. cerevisiae. We generated three distinct reporter libraries of the viral HRSVgp04 gene for studying the effect of three distinct regions in the transcript; (1) the 5'UTR, (2) the first 40 codons, and (3) codons 42-81 of the ORF. Each of the three libraries contained variants with multiple, rationally designed synonymous mutations, totaling 383 distinct variants tested individually for gene expression. Our results show that while synonymous mutations in each of the three regions can have a dramatic effect on protein abundance, those closer to the 5’end of the ORF are the most effective modulators of protein abundance. Additionally, while weaker local mRNA folding at the beginning of the ORF (codons 1-8) increases protein abundance, it decreases protein abundance when present in downstream codons, reinforcing previous evolutionary studies demonstrating the selection of folding strength in different parts of the ORF. Finally, we show that the mean relative codon decoding time, based on ribosomal densities in endogenous genes, significantly correlates with our measured protein abundance (correlation up to r = 0.6175; p=0.0013). While this report provides an improved understanding of transcript evolution and gene expression regulation, it also suggests relatively simple rules for engineering synthetic gene expression in a eukaryote.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
A canonical correlation analysis based dynamic Bayesian network prior to infer gene regulatory networks from multiple types of biological data
Brittany Baur1, Serdar Bozdag1
1Marquette University
One of the challenging and important computational problems in systems biology is to infer gene regulatory networks of biological systems. Several methods that exploit gene expression data have been developed to tackle this problem. In this study, we propose the use of copy number and DNA methylation data to infer gene regulatory networks. We developed an algorithm that scores regulatory interactions between genes based on canonical correlation analysis. In this algorithm, copy number or DNA methylation variables are treated as potential regulator variables and expression variables are treated as potential target variables. We first validated that the canonical correlation analysis method is able to infer true interactions in high accuracy. We showed that the use of DNA methylation or copy number datasets leads to improved inference over steady-state expression. Our results also showed that epigenetic and structural information could be used to infer directionality of regulatory interactions. Additional improvements in gene regulatory network inference can be gleaned from incorporating the result in an informative prior in a dynamic Bayesian algorithm. This is the first study that incorporates copy number and DNA methylation into an informative prior in dynamic Bayesian framework. By closely examining top-scoring interactions with different sources of epigenetic or structural information, we also identified potential novel regulatory interactions.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Disease gene prioritization using network and feature
Bingqing Xie1, Gady Agam1, Sandhya Balasubramanian2, Jinbo Xu3, Natalia Maltsev2, Conrad Gilliam2, Daniela Boernigen2
1Illinois Institute of Technology, 2University of Chicago, 3Toyota Technological Institute of Chicago
Identification of the most promising candidate genes contributing to the disease phenotypes among large lists of variations produced by high-throughput genomics using traditional experimental methods is time- and cost- consuming. Therefore, using computational approaches utilizing existing biological knowledge for the prioritization of such candidate genes will allow enhancing the efficiency and accuracy of the analysis of biomedical data. It will also allow reducing the cost of the studies by avoiding experimental validations of irrelevant candidates. To prioritize candidate genes contributing to a disease or phenotype of user’s interest for further testing, in this study, we present a novel algorithm that utilizes both types of information sources, gene annotations and gene interactions simultaneously, while preserving their original representation using Conditional Random Field (CRF) model. We further improve the accuracy and efficiency of our proposed approach by assigning enrichment scores to the annotation feature factors within the model. To estimate the performance of our approach, we evaluated it on two independent benchmark studies, ranking the candidate genes by both network and feature knowledge. Our results overall had high Area Under Curve (AUC) values and high partial AUC (pAUC) values on various diseases benchmarks and revealed a higher accuracy and precision at the top predictions (10%) as compared with other prioritization tools. Additionally, we applied our method on a case study for the prediction of molecular mechanisms contributing to intellectual disability and autism. Our method was able to recover additional genes related to both disorders and provide suggestions for possible candidates based on their rankings and functional categories.
top
Updated Nov 3, 2014
Links within this page:
...............................................................................................................................
Rheumatoid Arthritis Responder Challenge Posters
...............................................................................................................................
DREAM P01: A generic method for predicting clinical outcomes and drug response and its application in the RA challenge
Fan Zhu1 and Yuanfang Guan1,2,3
1University of Michigan
We developed an elegant Gaussian Process Regression (GPR)-based model to predict clinical outcomes and drug response. We applied it in both the genetics-only task and the genetics + clinical information-combined task in the DREAM Rheumatoid Arthritis Outcome Responder Challenge. It achieved the top accuracy in both the leaderboard and the final previously unseen test set, for both change in disease severity ( DAS) and predictions of non-response to treatment. We will present the rationale and method of this approach and elaborate the techniques of using it in predicting RA outcomes.
...............................................................................................................................
DREAM P02: Predicting response to arthritis treatments: regression-based Gaussian processes on small sets of SNPs
Javier García-García1 , Daniel Aguilar1, Daniel Poglayen1, Jaume Bonet1, Oriol Fornés1, Emre Güney2, Joan Planas-Iglesias1,3, Manuel Alejandro Marín1, Bernat Anton1 and Baldo Oliva1
1Universitat Pompeu Fabra, 2Northeastern University, 3Dana-Farber Cancer Institute, 4Current Address: University of Warwick
The aim of our study was to identify candidate SNPs playing a role in the response to therapy in rheumatoid arthritis (RA) patients, by compiling several sources of information such as the localization in the coding/non-coding region of the gene and its consequences in the translated protein (i.e. a synonym or non-synonym mutation). Genes affected by SNPs were first analyzed in order to select the most relevant associations with RA as follows. An initial list of potential candidates was selected using association analysis derived from the experimental data provided by the DREAM challenge. Additionally, we used multiple external sources of biomedical data to filter candidate SNPs. The list of candidates was expanded using gene-prioritization algorithms that combined protein-protein interaction networks and expression data. The procedure is based on the guilt-by-association principle and we selected from the extended list only those candidates with known SNPs. After the selection of genes, we used all SNPs reported for these genes. The resulting SNPs, in combination with clinical data, were used to predict the patients' response to treatments by means of regression-based models and a 10-fold cross-validation on the training dataset provided by the DREAM-challenge. When models were applied to an independent dataset (the leaderboard set), their predictive power decreased significantly, pointing out a problem of overfitting in the model. After comparison of the initial list of potential candidates and the use of external sources of information (i.e. biomedical data to filter the candidate list and extending the list using guilt-by-association principles), we confirmed that the predictive value of the original list of candidate SNPs was not improved by any of the external information. Therefore, we simplified the approach and reduced the SNP list by selecting only those showing the highest Pearson's correlation with the patients' response (DAS) in the leaderboard set (only about 20% of the initial SNPs). In the final independent dataset (CORRONA dataset) we achieved an AUC-ROC value of 0.6237 and AUC-PR value of 0.5071.
...............................................................................................................................
DREAM P03: Integrating prior biological knowledge into machine learning models for predicting drug responses
Lu Cheng 1,2,3 , Gopal Peddinti1, Muhammad Ammad-ud-din2,3, Alok Jaiswal1, Himanshu Chheda1, Suleiman Ali Khan1,2,3 , Kerstin Bunte2,3, Jing Tang1, Matti Pirinen1, Pekka Marttinen2,3, Janna Saarela1, Jukka Corander 2,4, Krister Wennerberg1, Samuel Kaski2,3, Tero Aittokallio1
1Institute for Molecular Medicine Finland (FIMM), University of Helsinki, 2Helsinki Institute for Information Technology (HIIT), 3 Aalto University, 4University of Helsinki
For complex genetic diseases such as rheumatoid arthritis, treatment effects can vary significantly among different patients. We integrate prior biological knowledge into machine learning models to explain the differences. We use GWAS, CCA based selection, PharmGKB, differential gene expression analyses to select the SNPs. We use GEMMA (linear mixture model) and BEMKL (multiple kernel methods) for our predictions. Our results show that the clinical information contributes the most for explaining the differences. Genetic information contributes a relatively small amount for the predictions, which is validated by comparing with clinical only predictions and random SNP set predictions. Correctly utilizing the methods is also very important. Our results show that changing the settings within these methods can create huge differences in predictions.
...............................................................................................................................
DREAM P04: DREAM Rheumatoid Arthritis Responder Challenge: Team Lucia
Victor Bellón 1,2,3 , Chloé-Agathe Azencott1,2,3, Véronique Stoven1,2,3, Olivier Collier1,2,3, Azadeh Khaleghi1,2,3, Valentina Boeva1,2,3 and Jean Philippe Vert1,2,3
1MINES ParisTech, PSL-Research University, 2Institut Curie, 3INSERM U900
Approximately 30% of rheumatoid arthritis patients do not respond to their treatment. In this challenge the focus was on anti-TNFα drugs. TNFα is involved in the inflammation pathway, which takes an important role in the disease. We use 2.5 million SNPs and simple clinical variables to predict the response of patients to their anti-TNFα drugs. We selected SNPs based on biological knowledge and their statistical relevance according to mutual information criteria. After selecting the SNPs, we use kernel methods to deal with the high dimensionality of the data. During the first phase of the challenge, we observed that genetic information is explaining only a small part of the data. Our team achieved the second best score in the first sub-challenge using only clinical variables.
Top of Page
ICGC-TCGA-DREAM Somatic Mutation Calling Challenge Posters
...............................................................................................................................
DREAM P05: novoBreak: robust characterization of structural breakpoints in cancer genomes
Zechen Chong1 , Ken Chen1
1University of Texas MD Anderson Cancer Center
Structural variation (SV) is a major source of genomic variation and plays a driving role in cancer genome evolution. However, the current strategy of using next-generation whole genome sequencing still does not achieve the comprehensiveness and sensitivity required to identify abundant SV breakpoints in heterogeneous tumor samples. This is due to challenges in acquiring high sequencing depth as well as methodological limitations in aligning and interpreting short reads spanning breakpoints. To alleviate such challenges and to deepen our understanding of cancer genome evolution, we developed a novel algorithm, novoBreak, which targets the reads that substantially differ from the normal genome reference and outputs the "breakome": the collection of genomic sequences spanning breakpoints and unobserved in the reference alignment. novoBreak can comprehensively characterize a variety of breakpoints that are introduced by small indels, large deletions, duplications, inversions, insertions and translocations at base-pair resolution from whole genome sequencing data. In contrast to most existing SV discovery programs such as Delly and Meerkat, novoBreak first clusters reads around potential breakpoints and then locally assembles the reads associated with each breakpoint into contigs. After aligning the contigs to the reference, novoBreak then identifies the precise breakpoints and infers the types of SVs. novoBreak performs substantively better than other widely used algorithms and ranked at No. 1 in the recent ICGC-TCGA DREAM Somatic Mutation Calling Challenge. The higher sensitivity of novoBreak makes it possible to uncover a large number of novel and rare SVs, as shown in our data from The Tumor Genome Atlas (TCGA) and from the 1000 Genomes project. Wider application of novoBreak is under way and is expected to definitively reveal the comprehensive structural landscape that can be linked to novel mechanistic signatures in cancer genomes.
...............................................................................................................................
DREAM P06: Applying logistic regression to combine multiple somatic mutation call sets for increased overall prediction accuracy
Li Tai Fang1,* , Pegah T. Afshar2,*, John C. Mu1, Narges Bani Asadi1, Wing H. Wong2,3, Hugo Y.K. Lam1
*Authors contributed equally
1Bina Technologies, 2Stanford University, 3Stanford University School of Medicine
Integrating multiple different algorithms to detect genetic variants or pathogenicity has been shown to be an effective approach in increasing the accuracy of prediction. It is worth noticing that multiple prediction algorithms are not independent lines of evidence, as pointed out by MacArthur et al. lately. Therefore, proper understanding and handling of the underlying algorithms being implemented are required to leverage the strengths of different algorithms and avoid mistreatment. Recent reviews comparing somatic mutation callers by Wang et al. and Roberts et al. have clearly shown that various well-established tools indeed have drastically contrasting performance in different situations such as varying allele frequencies. This indicates that integrating multiple algorithms can increase the sensitivity of detection, leaving the difficult question of how to maintain the specificity at an acceptable rate.
Previous attempts such as the pipeline Cake by Rashid et al. has taken the ensemble approach to provide an integrated analysis of somatic variants based on multiple callers with a series of post processing filters. However, it does not have a robust model that is proven to maximize the accuracy for any combination of tools. In this regard, Kim et al. have proposed a statistical model to combine calls from multiple somatic mutation callers based on regularized logistic regression with feature-weighted linear stacking (FWLS). The model was able to build a combined caller across the full range of stringency levels, which outperformed all of the individual ones. Based on this approach, we have carefully chosen four somatic callers, namely MuTect, VarScan2, JointSNVMix2, and SomaticSniper, based on their performance and characteristics. We combined their call sets with over 75 caller and sequencing features. Our approach is able to achieve high accuracy as demonstrated in the ICGC-TCGA DREAM Somatic Mutation Calling Challenge (the Challenge).
The four chosen algorithms compensate each other with their private calls in different circumstances. For example, MuTect is very sensitive at detecting low allele frequency mutations, but SomaticSniper and VarScan2 are more tolerant of calling candidates with mutation evidence in the matched normal. However, each tool also brings in extra false positive calls. Our approach improves precision with features that are predictive of variant calling confidence, such as the depth of coverage, alternate allele frequency, strand bias, mapping score, base call quality, and others. The final confidence score of each mutation candidate is the weighted sum of all the features. We applied our algorithm on the SNVs detected by the four callers from the synthetic reads in Stage 4 of the Challenge. From the union of the four call sets, it captured 78.7% of the total true mutations (sensitivity), but only 23.4% of them are true positives (precision). When our model was trained on Stage 3 data, we were able to improve the precision from 23.4% to 97.1%, while maintaining the sensitivity at 74.0%, achieving an accuracy (average of sensitivity and precision as computed in the Challenge) of 85.5%.
If we use Stage 4 dataset for cross-validation (i.e., randomly partitioning half of the stage 4 data for training, and the other half for validation), where we expect a high degree of consistency across the training and validation datasets, the accuracy is improved to 88.2%, with a sensitivity of 77.8% and precision of 98.6%. We demonstrated that using our machine learning approach with multiple call sets dramatically improves both sensitivity and specificity upon any single call set, even though sensitivity is capped at the combined performance. As a result, we envision that by incorporating more algorithmically different tools, our approach is able to achieve ultra-high accuracy.
References
1. Kim SY et al., Combining calls from multiple somatic mutation-callers, BMC Bioinformatics. 2014, 15:154. (doi:10.1186/1471-2105-15-154)
2. MacArthur DG et al., Guidelines for investigating causality of sequence variants in human disease, Nature. 2014, 508(7497):469-76. (doi: 10.1038/nature13127)
3. Roberts ND et al., A comparative analysis of algorithms for somatic SNV detection in cancer, Bioinformatics. 2013, 29(18):2223-30. (doi: 10.1093/bioinformatics/btt375)
4. Rashid M et al., Cake: a bioinformatics pipeline for the integrated analysis of somatic variants in cancer genomes. Bioinformatics. 2013, 29(17):2208-10. (doi: 10.1093/bioinformatics/btt371)
...............................................................................................................................
DREAM P07: Application of MuTect for sensitive and specific somatic point mutation detection in DREAM challenge synthetic data
Mara Rosenberg1, Kristian Cibulskis1, Adam Kiezun1, Louis Bergelson1, Gad Getz1,2
1Broad Institute of Harvard and MIT, 2Massachusetts General Hospital
Sensitive and specific detection of somatic point substitutions is a critical aspect of characterizing the cancer genome. However, tumor heterogeneity, purity, and sequencing errors confound the confident identification of events at low allelic fractions. MuTect, a previously described method for somatic mutation calling [1], allows for high sensitivity by first implementing a Bayesian classifier and then further reducing the false positives through carefully tuned filters. We applied MuTect to the four synthetic datasets in the DREAM challenge and achieved top scoring performance with specificity ranging from 0.98 to 0.99 and sensitivity from 0.74 to 0.97, consistent with our experience with real data. This had a corresponding false positive rate between 0.01 and 0.07 mutations per Mb. Here, we will describe our approach that used an application of MuTect and filters to reduce artifacts from bam alignment errors and base specific sequencing noise.
Reference
1. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213-9.
Top of Page
DREAM9 Acute Myeloid Leukemia (AML) Outcome Prediction Challenge Posters
...............................................................................................................................
DREAM P08: Evolution-informed modeling to predict AML outcomes
Li Liu1
1 Arizona State University
As a part of the DREAM9 Challenge, the Acute Myeloid Leukemia (AML) Outcome Prediction Subchallenge 1 aims to foretell if an AML patient will have a complete response or resistance to treatment based on 40 clinical covariates and 231 proteomic measurements. Previous analysis performed by the challenge organizers showed that the high level of noise in proteomic data reduced their predictive power when used in an uninformed manner. To solve this problem, I designed an evolution-informed model that incorporates weights derived from evolutionary conservation and univariate analysis in machine learning algorithms.
Based on evolutionary patterns of cancer genes, it can be inferred that changes of expression levels may have more profound impact if they involve conserved proteins, as compared to variable proteins. Therefore, higher weights can be given to slow-evolving proteins, and to proteins differentially expressed between two outcome groups. To estimate protein conservation, evolutionary rate (r) for each position in each protein was calculated based on alignments of orthologous sequences from 46 vertebrates. The evolutionary weight (WE) of a protein was the reciprocal of average evolutionary rate over all positions ( 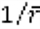 ). Clinical variables took the maximum of all WEs. Next, Student's t-test was performed for each feature. P-values were transformed via negative logarithm (-log(P)) and used as the differential weight (WD). For a given feature, the final weight was the sum of its evolutionary and differential weights (W = WE + WD).
). Clinical variables took the maximum of all WEs. Next, Student's t-test was performed for each feature. P-values were transformed via negative logarithm (-log(P)) and used as the differential weight (WD). For a given feature, the final weight was the sum of its evolutionary and differential weights (W = WE + WD).
In the feature selection step, each feature was first transformed to z scores, and then multiplied with its corresponding weight (W). Because the training data were highly unbalanced, an ensemble approach was used to construct multiple classification models with balanced subsamples. Using stability selection with sparse logistic regression, features identified in >50% of bootstrapping runs were selected. In the classification step, these features with un-weighed values were used to construct a random forest model with 50 trees. The above procedure was repeated 100 times to produce an ensemble of 100 random forest models. Given a patient, 100 predictions were obtained, one from each model. The confidence score equals the proportion of models that predict the patient to have a complete response.
When applied to test data that are blind to participants of this challenge, this evolution-informed model achieved a balanced accuracy of 77.9% and AUROC of 0.795, ranked number one among all participants. Features that were selected in more than 80% of all models include Chemo (Flu-HDAC), cyto.cat (21), CD34, cyto.cat (-5), Age.at.Dx, ABS.BLST, PIK3CA, and GSKA_B.pS21_9.
...............................................................................................................................
DREAM P09: Acute myeloid leukemia outcome prediction via dictionary learning for sparse coding
Zhilin Yang1, Subarna Sinha2, David L. Dill2
1 Tsinghua University, 2Stanford University
This challenge was to use clinical and reverse-phase protein array (RPPA) data to solve three subchallenges: predicting complete remission after treatment, predicting remission duration, and predicting survival time. We describe our solutions to the first two subchallenges. For the first challenge, we found that a support vector machine (SVM) classifier with the radial basis function kernel was the most effective standard classifier of those we tried. We added a manual rule that any patient treated with Flu-HDAC would experience remission.
We found it difficult to improve prediction performance using RPPA data until we used dictionary learning for sparse coding, which learns low-rank latent state vectors from the original data in an unsupervised way and represents each sample as a sparse linear combination of the latent states, for feature extraction. Using sparse coding features of all protein data improved classifier performance. Applying sparse coding to pathway-specific subsets of proteins improved performance further, showing that prior knowledge of pathways can be useful in this task. Interestingly, some of the latent states in the pathway-specific sparse codes seemed to be biologically meaningful. The quality of the results also depended on a hybrid feature selection algorithm for clinical variables to avoid mixing up continuous and categorical features. We observed significant batch effect in the RPPA data, which we tried to correct unsuccessfully using several standard methods.
For the second subchallenge, we used an average of three support vector regressions using different subsets of the features. We were unable to improve the quality of predictions in this subchallenge using the RPPA data.
...............................................................................................................................
DREAM P10: A bagged, semi-parametric model to predict survival time for acute myeloid leukemia patients
Xihui Lin1 , Gregory M. Chen1, Honglei Xie1, Geoffrey A. M. Hunter1, Paul C. Boutros1,2
1Ontario Institute for Cancer Research, 2University of Toronto
While many AML patients go into remission after treatment, survival time remains highly variable across individuals. Predicting these differences would be of major clinical value in personalizing therapy. As part of the ninth Dialogue for Reverse Engineering Assessment and M ethods (DREAM9) challenge, we sought to accurately estimate the survival of AML patients by integrating clinical and proteomic features. We initially formulated survival models based on random forests, boosted quantile regression, and weighted linear models, but these performed no better than a benchmark Cox model with only five clinical variables. Therefore, we decided to extend the benchmark Cox model for our final submission in the DREAM9 challenge. Specifically, we used a bootstrap aggregated (bagged) modified Cox model based on five clinical features: age at diagnosis, Anthra based treatment administered, hemoglobin count, Albumin levels, and cytogenic category. Researchers identified cytogenics as the single most important prognostic factor in AML patients; however, the cytogenic categories in the data for the challenge were imbalanced. To resolve this, we re-stratified patients into high, intermediate, intermediate-low, and low risk survival groups based on their cytogenic category. This significantly improved the predictive power of the model. Surprisingly, incorporating additional clinical and/or proteomic features in the Cox model diminished its performance. These results suggest that our reclassified cytogenic categories can improve predictions of patient survival and, hence, might be the key to help tailor therapies for AML patients.
...............................................................................................................................
DREAM P11: Predicting overall survival of AML patients with Cox proportional hazards model
Ljubomir Buturovic1 , Damjan Krstajic1,2,3
1Clinical Persona Inc., 2Research Centre for Cheminformatics, Belgrade3University of Belgrade
One of the goals of the AML Challenge was to predict Overall Survival of AML patients using clinical and proteomics data. We tried the following two approaches to find an optimal survival model:
- Cox proportional hazards model
- parametric survival model
Bovelstad et al. [1] built various survival models on clinical and genomics data and showed that on average, a ridge-regularized Cox proportional hazards model outperforms others. However, it is not ideally suited for predicting actual survival (expected time until death). The reason is that baseline hazard in the Cox model is unknown, which made it difficult to apply in the overall survival subchallenge. However, the strength of a Cox model is that it may provide risk scores for various time points. To overcome the baseline hazard issue, we used 600 * S(600) as the measure for predicting overall survival in Cox models, where S(t) is the patient's survival function estimated by the Cox model, and 600 is the maximum observed survival time in the subchallenge.
Parametric survival models are more suited for predicting overall survival, because their baseline hazard is well defined. However, their main weakness is that they usually overfit.
As described in our recent publication [2], we used the grid-search repeated cross-validation approach to select an optimal model. The best cross-validation results were found for regularized Cox regression model using the glmnet R package. Our results confirm the findings by Bovelstad et al. [1] that regularized Cox regression models are serious contenders for clinico-genomic survival models.
References
1. Bovelstad HM, Nygard S, Borgan O. Survival prediction from clinico-genomic models - a comparative study. BMC Bioinformatics 2009, 10:413.
2. Krstajic D, Buturovic LJ, Leahy DE, Thomas S. (2014). Cross-validation pitfalls when selecting and assessing regression and classification models. Journal of cheminformatics, 6(1), 1-15.
...............................................................................................................................
DREAM P12: Ensembling classical supervised statistical learning methods and survival models in the DREAM9 AML Challenge
Rolland He 1
1Stanford University
The DREAM9 AML Challenge provides a novel training dataset detailing the clinical covariates and proteomic measurements of 191 patients diagnosed with acute myeloid leukemia (AML). This dataset provides the basis for applications of conventional supervised statistical learning methods, including linear elastic net regression and gradient boosting machines, as well as the well-known Cox survival model. Despite being relatively simple to implement and understand, these models prove to provide competitive results when combined together. I will discuss some of the practical limitations of the dataset, some of the missteps I ran into along the way, as well as the implications of my results for future biostatistical analysis. In particular, it is important to understand the versatility of classical supervised statistical models and the strength of ensembling to form accurate predictions.
...............................................................................................................................
DREAM P13: Application of crowd sourcing to improve predictive models of AML outcome: constructing the DREAM 9 AML challenge
David Noren1 , Byron Long1, Raquel Norel2, Gustavo Stolovitzky2, Steven Kornblau3, Amina Qutub1
1Rice University, 2IBM Computational Biology Center, 3University of Texas MD Anderson Cancer Center
It is projected that clinical informatics will have a profound impact on the immediate future of health care. In particular, insights drawn from clinical genomic, proteomic, and metabolomic data have the potential to greatly improve the accuracy of patient prognosis and the effectiveness of therapeutic selection. However, to realize this potential, there is a need to explore, develop, and validate new computational algorithms that are capable of translating high throughput measurements into clinically useful information. The Dialog on Reverse Engineering Assessments and Methods (DREAM) is a crowd sourcing effort which has pioneered the competitive and cooperative development of predictive biological models to fill this need. Each DREAM endeavor is structured around a challenge framework where participants from different technical fields are invited to contribute their best solutions. Here we describe the background, design, and implementation of the DREAM 9 Acute Myeloid Leukemia (AML) Outcome Prediction Challenge.
Acute myeloid leukemia (AML) is a devastating cancer of the bone marrow and the blood. It is predicted that there will be over 18,860 new cases this year and 10,460 deaths attributed to AML. While information pertaining to patient mutation status and cytogenetics have assisted clinicians in matching different therapies to specific subsets of patients, the effectiveness of treatment remains low and the overall 5 year survival rate is only ~25%. To improve personalized therapy for these patients, MD Anderson Cancer Center has employed new proteomic technologies, like Reverse Phase Protein Array (RPPA). RPPA allows clinicians to directly ascertain the changes in AML patient signaling proteins, which are the molecular targets of most therapeutics. The goal of the DREAM9 AML Outcome Prediction Challenge is to develop models that are predictive of patient outcome using the RPPA proteomics data in conjunction with other clinical measurements.
The design of the DREAM9 AML Outcome Prediction Challenge embodied several components, each with special considerations. Clinical data is often "messy" and thus the AML data was first processed to insure clarity and consistency. Two separate datasets were identified, a training dataset that was given to participants to develop their models and a test set that was later used to evaluate participant predictions. These datasets were chosen to insure the training data provided an adequate representation of the overall dataset. Different scoring metrics were evaluated to measure participant performance as well as to evaluate the threshold signal to noise ratio. Finally, a feedback structure was also designed - i.e., leaderboard schedule - to assist participants with model development without promoting model over-fitting. Here we describe the methodology and tools used to address each of these considerations when designing the DREAM9 AML Outcome Prediction Challenge and review some of the preliminary results.
Top of Page
Broad-DREAM Gene Essentiality Prediction Challenge Posters
...............................................................................................................................
DREAM P14: Learning kernel-based feature representation for gene essentiality prediction
Masayuki Karasuyama 1 , Hiroshi Mamitsuka1
1Kyoto University
We develop a predictive method for estimating gene essentiality, focusing on learning a predictive feature representation. Our method uses a kernel technique, in which the kernel is trained to capture mutual relations among different cell-lines, with respect to essentiality. We start with our baseline model, kernel ridge regression (KRR), a well-known, stably high-predictive performance model. We then attempt to improve the predictive performance of KRR by learning the kernel (or features) from gene essentiality data itself. More concretely we focus on the essentiality scores of genes, in given data, where the scores of different genes are heavily dependent on each other. We incorporate this dependency into our predictive model by using kernel canonical correlation analysis (KCCA) and kernel target alignment (KTA), both of which can be interpreted as estimating feature representations using the 'ideal' kernel defined by essentiality scores. After obtaining kernels through KCCA and KTA, we then predict the essentiality of an arbitrary gene by using the two KRR models. We finally take the average over the two prediction results (by the two models) to stabilize the results. An important point of our model is that the trained kernel is shared with all genes to predict the essentiality of each gene. This point reduces estimation variance, which can be a severe problem in high dimensional and small sample data (which is applied to the given data this time), rather than estimating different kernels for each gene. Overall, these modifications make our predictive model a high-performance approach, particularly in subchallenge 1 of the Broad-DREAM Gene Essentiality Prediction Challenge. An additional, big advantage of our approach is computational efficiency, because all techniques (KCCA, KTA and KRR) used in our approach are kernel methods, in which we do not have to deal with high dimensional data directly after we once calculate the kernels.
...............................................................................................................................
DREAM P15: Multi Pathway Learning accurately predicts gene essentiality in the Cancer Cell Line Encyclopedia
Vladislav Uzunangelov 1 , Sahil Chopra1, Kiley Graim1, Daniel Carlin1,Yulia Newton1, Alden Deran1, Adrian Bivol1, Sam Ng1, Kyle Ellrott1, Joshua M. Stuart1+, Artem Sokolov1+ and Evan Paull1+.
1University of California, Santa Cruz
+Corresponding author(s): This email address is being protected from spambots. You need JavaScript enabled to view it., This email address is being protected from spambots. You need JavaScript enabled to view it., This email address is being protected from spambots. You need JavaScript enabled to view it.
We applied biologically motivated feature transformations coupled with established machine learning methods to predict gene essentiality in CCLE cell line models. By leveraging additional large datasets, such as The Cancer Genome Atlas PanCancer12 data and MSigDB pathway definitions, we improved the robustness and biological interpretability of our models. We developed a multi-pathway learning (MPL) approach that associates a genetic pathway from MSigDB with a distinct kernel for use in a multiple kernel learning setting. We evaluated the performance of MPL compared to several other regression methods including random forests, kernel ridge regression, and elastic net linear models. We combined multiple approaches using an ensemble technique on the diverse set of predictors.
We found that the winning method was an ensemble that combined the random forest and MPL predictions. Both models utilized features derived from both gene expression and copy number data, the latter of which were filtered to those predicted as driver events in prior pan-cancer studies. MPL also ranked as the 5th highest performing method on the third sub-challenge. In this case, the 100 features with the highest cumulative MPL weights were selected and applied with kernel ridge regression to produce the final predictions. Thus, MPL also demonstrates merit as a feature selector when used with other downstream methods.
The method performed the best at predicting essentiality of those genes belonging to classes such as kinases (31 out of top 100), fibronectin type III (7 out of 100), and insulin signaling (6 out of top 100). Kinases represent a broad class of genes whose knock-outs are expected to have a wide range of effects due to their master regulatory role. Thus, signatures of gene essentiality for these genes might be more readily inferred. In addition, high prediction accuracy was achieved for several genes involved in cancer, such as TP53, PIK3CA, RB1, FGFR1, ABL1, and FLT3, suggesting MPL's utility as a biomarker for detecting key tumorigenic events.
The advantage of MPL is that mechanistically coherent gene sets are automatically selected as high scoring pathway kernels (HSPKs). We investigated whether the HSPKs identify cellular processes relevant to the loss of key genes. To do this, we inspected the HSPKs for a few of the most abundantly mutated genes in cancer. The MPL predictor for TP53 included the targets of this transcription factor as well as HSPKs involved in apoptosis, a cellular process regulated by TP53. The retinoblastoma gene (RB1) MPL predictor included RB1 targets as well as HSPKs involved in the regulation of histone deacetylase (HDAC) that interacts with RB1 to suppress DNA synthesis. PIK3CA, a gene that is mutated frequently in luminal, but not basal, breast cancers was associated with HSPKs comprised of genes differentially expressed in luminal breast cancers. Finally, HSPKs predictive of BRAF essentiality included genes associated with uveal melanoma, a finding consistent with the prevalence of BRAF mutations in skin cancers. These findings suggest trends in the MPL results could reveal a pathway-level view of the synthetic lethal architecture of cells. Such a map, that links patterns of pathway expression to potential genetic vulnerabilities, could provide an invaluable tool for exploring new avenues to target cancer cells.
...............................................................................................................................
DREAM P16: Integrative model to predict gene essentiality for cancer cell survival
Tao Wang1, Xiaowei Zhan1, Hao Tang1, Yang Xie1, Guanghua Xiao1
1University of Texas Southwestern Medical Center
The central question of precision medicine in cancer is how to identify patients who are more susceptible to a given treatment. This is especially important for targeted cancer therapies, which target specific genes/pathways to inhibit cancer cell growth and may only be effective for a specific sub-population. It is critical to predict the extent to which cell survival relies on specific genes (gene essentiality), which are the target genes for cancer treatment. Here, we show that an advanced data-driven dimension reduction strategy of integrating features from expression, copy number, and mutation data, coupled with Gaussian Process for Regression (GPR), is an effective approach to predicting gene essentiality. The feature selection step takes several factors into consideration, including the gene expression level, the distribution difference between the training and testing data, and the association with outcome variables. We applied GPR on the selected features and specified appropriate kernels to capture the complex non-linear relationship between predictors and response. In conclusion, we developed a best-performing predictive pipeline to solve the Broad-DREAM Gene Essentiality Prediction challenge (sub-challenge 1) and provided new perspectives for the realization of precision medicine in cancer.
...............................................................................................................................
DREAM P17: A strategy to select most informative biomarkers for cancer cell lines
Fan Zhu 1 , Yuanfang Guan1
1 University of Michigan
Cancer cells represent strong heterogeneity and thus the response to treatment varies dramatically between individuals. Currently a rough estimation of 80% of the patients do not respond to cancer therapy. Personalized treatment of tumors thus requires accurate identification of drug targets for the specific samples collected from biopsy. Ideally, a test panel with a limited number of biomarkers can be designed for each type of cancer to identify effective drug targets for a patient. The Broad Institute Gene Essentiality Subchallenge 2 studies whether such biomarkers can be found for each type of cancer. We have developed a method to rigorously select such stable biomarkers based on both their informativeness in the cell line under investigation and the global informativeness over all cell lines. This was the best-performing method in this subchallenge.
...............................................................................................................................
DREAM P18: Predicting gene essentiality using linear-time greedy feature selection
Peddinti Gopalacharyulu*1, Alok Jaiswal*1, Kerstin Bunte2, Suleiman Khan1,2,Jing Tang1, Antti Airola 4, Krister Wennerberg1, Tapio Pahikkala4, Samuel Kaski2,3, Tero Aittokallio1
*Equal contributions
1Institute for Molecular Medicine Finland FIMM, University of Helsinki, 2Helsinki Institute for Information Technology HIIT, Aalto University, 3Helsinki Institute for Information Technology HIIT, University of Helsinki, 4University of Turku
Genome-wide prediction of the gene essentiality using molecular characteristics of various cancer cells has the potential to open up new avenues for selective cancer therapies as well as for providing insights into the systems-level genetic interaction networks of cancer cells. Subchallenges 2 and 3 of the Broad-DREAM9 Gene Essentiality Prediction Challenge deal with the problem of finding a limited number of molecular features that are most predictive of gene essentiality. To solve this problem, we used a greedy forward feature selection algorithm for regularized least squares (RLS), called GreedyRLS. The GreedyRLS algorithm works like a wrapper type of feature selection method, which starts with an empty feature set, and in each iteration adds the feature whose addition provides the minimum RLS error in the leave-one-out cross-validation (LOO-CV). The GreedyRLS algorithm, however, performs the feature selection computationally more efficiently than previously known feature selection algorithms for RLS. The time complexity of the standard approach using LOO-CV for forward selection of k features from a total number of n features in a data set with m training samples is 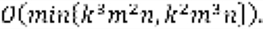 In contrast, the time complexity of the GreedyRLS is
In contrast, the time complexity of the GreedyRLS is 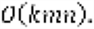 In sub-challenge 3, we utilized the GreedyRLS approach for multi-task learning, and it performed the best among all the competing methods in this sub-challenge. We addressed subchallenge 1 using additional information based on pathways from PARADIGM and gene sets from MSigDB. In this subchallenge, we used the Bayesian multitask multiple kernel learning (BEMKL) method, which is a non-linear method based on kernelized regression and Bayesian inference. Use of additional information of similarities of genes based on gene ontology seemed to be helpful in predicting gene essentiality, in line with the lessons learned from the previous NHI-DREAM Drug Sensitivity Prediction Challenge, but did not lead to the top performance in this subchallenge.
In sub-challenge 3, we utilized the GreedyRLS approach for multi-task learning, and it performed the best among all the competing methods in this sub-challenge. We addressed subchallenge 1 using additional information based on pathways from PARADIGM and gene sets from MSigDB. In this subchallenge, we used the Bayesian multitask multiple kernel learning (BEMKL) method, which is a non-linear method based on kernelized regression and Bayesian inference. Use of additional information of similarities of genes based on gene ontology seemed to be helpful in predicting gene essentiality, in line with the lessons learned from the previous NHI-DREAM Drug Sensitivity Prediction Challenge, but did not lead to the top performance in this subchallenge.
...............................................................................................................................
DREAM P19: Integrative learning of gene essentiality using data and knowledge with cluster specific models
Simone Rizzetto1 , Paurush Praveen1, Mario Lauria1, Corrado Priami1,2
1The Microsoft Research-University of Trento Centre for Computational and Systems Biology, 2 University of Trento
Motivation: Predictive models to infer gene essentiality in cancer cell lines can aid the molecular characterization of the cancer cell lines, which can be ultimately used to identify biomarkers and tailored treatments as well as identify patients with higher treatment efficacy. The approaches to predict essentiality have been based on genome scale data (Roberts et al. 2007) as well as on network of genes (Kim et al. 2012). However, the gene essentiality should be seen as a context based term or measurement. For example, a gene essential in lung cancer cell line may not be essential in a breast cancer cell line and vice versa. Therefore, a generalized model for quantitative estimation of gene essentiality across heterogeneous cell lines is not suitable. Another limitation of current approaches, even with gene-specific models, is that they tend to use data from heterogeneous or diverse cell lines rendering the overall model noisy and causing a reduction in their predictive power. Another area that has been less exploited is the use of existing information or knowledge on the genes as well as cell lines that can boost the performance of quantitative prediction models. We aim to exploit these hypotheses while solving the issue of essentiality prediction in the challenge (Broad-Dream Gene Essentiality Prediction Challenge-2014). We propose two models, (1) One Gene-One Model (OGOM) with integrated knowledge features and (2) Cluster Specific-OGOM (CS-OGOM) addressing these issues.
Methods: We developed two approaches to build a model for quantitative prediction of gene essentialities, based on expression and copy number variation (CNV) data. A Support Vector Regression (e-SVR) (Smola & Scholkopf 2004) forms the underlying engine for prediction. The key aspects of the models have been highlighted below.
1. OGOM (One Gene One Model): The model is based on training one model for each gene for all cell lines using support vector regression. The difference is made by the integration of knowledge from freely available multiple information sources (Oncogene, cell line information, etc.) as features in addition to the gene expression and CNV data provided by the organizers.
2. CS-OGOM (Cluster-Specific OGOM): The CS-OGOM approach is based on the hypothesis that closely related cell lines will follow one model for each gene in order to predict its essentiality. Thus, compared to the OGOM approach we have a gene specific model for every cluster. The approach first performs a hierarchical clustering on the expression data to identify closely related cell lines. To obtain an optimal cluster homogeneity we used a distance metrics, based on the degree of similarity between rank-based signatures (Lauria et al. 2013). Now within each cluster we identify the training and test data. The training data within that cluster is used to learn a model for each gene and use this model to predict the essentiality of the corresponding gene in the test cell line member of the corresponding cluster.
Results: We applied leave one out cross validation to both the models in order to test the performance of our models and measured the performance in terms of correlation coefficients. The results showed that both models outperformed the generalized model (one-model for all cell lines and genes). The CS-OGOM performed substantially better than the OGOM in terms of Pearson and Spearman's coefficients. The Pearson's correlation for the CS-OGOM was found to be > 0.4 on average whereas on the Spearman's scale it oscillated around 0.3 depending on the gene under observation. The score for the OGOM were ~ 0.2 along both the scale. The inclusion of prior knowledge also improved the performance of OGOM without these features.
Discussion: Our models revealed the improvement brought about by the gene-specificity in models and integration of prior knowledge. The leave one out cross validation of CS-OGOM showed that the similar cell lines when used to train a model lead to better performance for specific cell lines. However, the choice for the number of clusters needs a trade-off between specificity (cell line homogeneity) and number of training samples. Another important aspect is the scaling of training and test data in order to avoid numerical artifacts while mixing the clusters for the final performance measure.
References
1. Roberts SB, Mazurie AJ, Buck GA (2007) Integrating Genome-Scale Data for Gene Essentiality Prediction. Chemistry & Biodiversity 4: 2618-2630.
2. Kim J, Kim I, Han SK, Bowie JU, Kim S (2012) Network rewiring is an important mechanism of gene essentiality change. Sci Rep
3. Smola AJ, Scholkopf B (2004) A tutorial on support vector regression. Statistics and Computing 14:199-222.
4. Lauria M (2013) Rank-based transcriptional signatures: A novel approach to diagnostic biomarker definition and analysis. Systems Biomedicine 1: 228-239.
...............................................................................................................................
DREAM P20: 5Cell viability prediction from large-scale omics data using machine learning and mechanistic modeling approaches
Emanuel Gonçalves1, Daniel Machado2, Michael Menden1, Julio Saez-Rodriguez1, Miguel Rocha2
1 EMBL-EBI European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, CB10 1SD, Cambridge, UK
2 Centre of Biological Engineering, University of Minho, 4710-057 Braga, Portugal
Systematic studies of the impact of genes loss of function, such as short hairpin RNA (shRNA) or CRISPRs, in cancer cells viability helps to identify vulnerabilities that can be explored for possible therapeutic treatments.
A current challenge in this field is the accurate prediction of gene essentiality given a genomic context, which can be characterised with gene copy number variation, mutation profiles and transcriptomic expression data. In this context, the final submissions of our team, UM-EBI, to the DREAM 9 Gene Essentiality challenge were performed using a combination of machine learning methods. For feature selection, we discarded features behaving similarly across all the samples, and ranked the features using an univariate feature selection method. Regarding model estimation, we used a “wisdom of crowds” approach that consisted on averaging the results of different estimation methods, which included Ridge regression, support vector machines (SVM) and Passive Aggressive regression (PAR).
We observed that gene expression was the most informative data set. In many cases, considering copy-number variation and mutation data decreased the performance of the estimators. Changes in the prediction methods resulted in small perturbations to the final scores, whereas the filtering and selection steps significantly affected our predictions.
One limitation of these machine learning approaches is the lack of biological insight that can be obtained. Furthermore, they do not take into account the compendium of information that has already been assembled and curated into mechanistic models. In this regard, we propose to extend our approach by considering genome-scale models of human metabolism and their regulation. These models capture biochemical processes that affect cell growth and viability, allowing mechanistic and quantitative predictions of the impact of genetic changes into the cellular growth phenotype. Methods have been recently developed to generate tissue-specific models for human cells from gene expression data. We are extending these methods to generate models of metabolism, gene regulation and signal transduction by integration of multiple omics data, that we can plan to apply to study gene essentiality.
Top of Page
Updated Oct 23, 2014
App P01: PEPPER: Cytoscape app for protein complex expansion using protein-protein interaction networks
Charles Winterhalter1,2, Rémy Nicolle1,3, Anais Louis1, Cuong To1, François Radvanyi3, Mohamed Elati1
1University of Evry, 2Newcastle University, 3CNRS/Institut Curie
We present PEPPER (Winterhalter et al., 2014), a Cytoscape app designed to identify protein complexes as densely connected subnetworks from seed lists of proteins derived from proteomic studies. PEPPER identifies connected subgraph by using multi-objective optimization involving two functions: (i) the coverage, a solution must contain as many proteins from the seed as possible, (ii) the density, the proteins of a solution must be as connected as possible, using only interactions from a proteome-wide interaction network. Comparisons based on gold standard yeast and human datasets showed PEPPER’s integrative approach as superior to standard protein complex discovery methods. The visualization and interpretation of the results are facilitated by an automated post-processing pipeline based on topological analysis and data integration about the predicted complex proteins. PEPPER is a user-friendly tool that can be used to analyze any list of proteins. PEPPER is available from the Cytoscape plugin manager or online (http://apps.cytoscape.org/apps/pepper) and released under GNU General Public License version 3.
Reference:
C. Winterhalter, R. Nicolle, A. Louis, C. To, F. Radvanyi, and M. Elati
PEPPER: cytoscape app for protein complex expansion using protein–protein interaction networks. Bioinformatics, 2014 doi:10.1093/bioinformatics/btu517
........................................................................................................
App P02: cyREST: initial steps toward the Cytoscape Cyberinfrastructure
Keiichiro Ono1, Barry Demchak1
1University of California San Diego
The Cytoscape application (and apps) have proven effective in powering research in systems biology. However, modern applications require more diverse workflows, more flexible automation, and access to more forms of data than is commonly provided by Cytoscape alone. While Cytoscape specializes in network analysis, visualization, and publishing, other tools have evolved sophisticated workflow, automation, data acquisition, and analysis capabilities that are potentially complementary to Cytoscape. The most popular of these tools include integrated data analysis environments (e.g., IPython Notebook, Matlab, and RStudio), analysis toolkits (e.g., Bioconductor, pandas, and igraph), scripting languages (e.g., Python and R), and fully featured workflow managers (e.g., Taverna and BioKepler).
To deliver the benefits of integrating Cytoscape with best-of-breed tools, we are creating the Cytoscape Cyberinfrastructure (CI) as scalable Internet-based computing environment based on a service oriented architecture (SOA) approach. As such, it views computational units as loosely coupled services whose functionality is integrated into workflows. The integration of Cytoscape with new and existing analytical and workflow tools represents a first step.
To realize Cytoscape as a service, we introduce cyREST, a Cytoscape app that exposes Cytoscape network data structures, Visual Styles, network layouts, data, and network publishing functions as RESTful services. cyREST enables external tools to retrieve, update, create, and delete entire networks or individual network nodes, edges, and groups. It also enables tools to retrieve and update node and edge annotation data, to define and execute annotation-based filters, or to define annotation-based node and edge visual styles. Finally, cyREST allows tools to invoke any of Cytoscape’s 15 standard (non-yFiles) network layouts, and then retrieve layout information.
To ease integration with tools and other services, cyREST observes defacto standard REST name space and communications practices, including using URLs to name conceptual entities (instead of as commands), using the HTTP protocol, and using the JSON data format.
We have already seen interest in using Cytoscape services from within IPython Notebook and RStudio, where users combine layout services with native data collection and popular graph analysis packages such as igraph and NetworkX, and the entire analysis and visualization workflow can be saved as executable code which improves research reproducibility. Because cyREST exposes a Cytoscape-centric data model, we supply tool-specific interface libraries (for R and Python, so far) that harmonize tool data models with Cytoscape’s.
The Cytoscape CI will enable researchers to efficiently realize workflows leveraging the strongest features of each tool in a scalable, reusable, and flexible manner. The combination of Cytoscape/cyREST enables existing tools to avoid duplicating Cytoscape’s powerful network management, filtering, and layout features, thereby powering high productivity for systems biology researchers.
........................................................................................................
App P03: Integrated omics analysis pipeline for model organism with Cytoscape
Kozo Nishida1, Koichi Takahashi1
1RIKEN
Although Cytoscape is a powerful tool for biological data analysis and visualization, it is not the best tool for data preparation or cleansing. For biologists, R and Python are the most popular programming languages and if we provide data analysis and visualization pipelines written in those two languages, it improves reproducibility of the research. It was impossible to integrate Cytoscape in such pipelines, but recently cyREST (https://github.com/keiono/cy-rest) a Cytoscape app to provide RESTful API to access low-level Cytoscape data objects, has been announced. If we use it with IPython notebook or R Markdown, we can write an executable data analysis workflow as R/Python code.
To write the data analysis pipeline, we have developed a Cytoscape app called KEGGscape to import KEGG PATHWAY XML (KGML) files. KEGGscape utilizes the database to reproduce the corresponding hand-drawn pathway diagrams with as much detail as possible in Cytoscape. KEGGscape is included in the pipelines and users can visualize their datasets on KEGG.
With these tools, we compiled a workflow to integrate and visualize pathway data (e.g., KGML, BioPAX, SBML, drug targets) and omics-datasets on E. coli and A. thaliana pathways. As a pilot project, we integrated drug targets from Drugbank (http://www.drugbank.ca/), The E. Coli Metabolome Database (ECMDB), iJO1366 (metabolic network reconstruction), and microarray datasets available in Bioconductor with KEGG PATHWAY database for E. coli. In a similar way, for A. thaliana, transcriptome, metabolome and various pathway datasets from RIKEN are also integrated with KEGG pathways.
........................................................................................................
App P04: The Cyni framework for network inference in Cytoscape
Oriol Guitart-Pla1, Manjunath Kustagi2, Frank Rügheimer1, Andrea Califano2, Benno Schwikowski1
1Institut Pasteur, 2 Columbia University
Research on methods for the inference of networks from biological data is making significant advances, but the adoption of network inference in biomedical research practice is lagging behind. Here, we present Cyni, an open-source ‘fill-in-the-algorithm’ framework that provides common network inference functionality and user interface elements. Cyni allows the rapid transformation of Java-based network inference prototypes into apps of the popular open-source Cytoscape network analysis and visualization ecosystem. Merely placing the resulting app in the Cytoscape App store makes the method accessible to a worldwide community of biomedical researchers by mouse click. In a case study, we illustrate the transformation of an ARACNE implementation into a Cytoscape app.
........................................................................................................
App P05: WikiPathways App
Martina Kutmon1, Samad Lotia2, Chris Evelo1, Alexander Pico2
1Maastricht University, 2Gladstone Institutes
Here we present the open-source WikiPathways app for Cytoscape that can be used to import biological pathways for data visualization and network analysis. WikiPathways is an open, collaborative biological pathway database that provides fully annotated pathway diagrams for manual download or through web services. The WikiPathways app allows users to load pathways in two different views: as an annotated pathway ideal for data visualization and as a simple network to perform computational analysis. An example pathway and dataset are used to demonstrate the functionality of the WikiPathways app.
........................................................................................................
App P06: CyNDEx - Accessing NDEx from Cytoscape
Dexter Pratt1, David Welker1, Jing Chen1
1University of California San Diego
CyNDEx provides connectivity to NDEx servers, enabling users to find, query, download, and store networks. Public networks can be accessed anonymously but users who have previously created accounts on an NDEx server can access private networks to which they have access permission.
........................................................................................................
App P07: clusterMaker2’s “Fuzzifier”: a simple approach to fuzzy clustering for biological networks
John "Scooter" Morris1, Abhiraj Tomar2, Thomas Ferrin1
1University of California San Francisco, 2University of Southern California
Clustering has become a standard approach to analyzing biological networks, but so far, traditional clustering approaches for biological networks have focused on “hard” or discrete clusters that remove edges from the graph until some optimal fit is achieved. This poster describes a new post-clustering step that has been implemented in the Cytoscape App clusterMaker2 (http://apps.cytoscape.org/apps/clusterMaker2). This post clustering step utilizes the results from a discrete network partition algorithm to seed a “fuzzy” clustering of the same data to provide information about the marginal assignments to clusters. This approach allows the user to take advantage of many existing cluster algorithms that have demonstrated results for particular types of biological systems. In the stepwise example below, we show how additional information is derived from a discrete clustering (using MCL) of a well-known protein-protein interaction data set.
........................................................................................................
App P08: CRE Browser: visualizing causal hypotheses
Ranjit Randhawa1, Dmitri Bichko2, David Klatte2, Daniel Ziemek2
1Novartis Institutes for BioMedical Research, 2Pfizer Worldwide Research and Development
The Causal Reasoning Engine (CRE) [1] is a platform used to analyze experimental data in the context of prior biological knowledge, to generate testable hypotheses about the upstream molecular drivers of observed changes. The CRE Browser is an interactive analysis tool, developed as a Cytoscape plugin, to help scientists visualize and explore hypotheses generated by CRE. CRE Browser uses a graph-centric interface to provide both a comprehensible view of the entire result set, and detailed supporting evidence for individual hypotheses.
CRE Browser comprises several components:
• Overview Graph shows the global view of the candidate hypothesis interactions, and allows users to cluster, filter, and explore the network topology to narrow in on the causal events of interest.
• Hypothesis View provides a deep-dive on an individual hypothesis, its structure, and underlying evidence. This view also includes a number of additional visualizations for the hypothesis statistics and associated experimental data, and services to integrate external data, such as literature mining results.
• Multi-Experiment View brings together hypothesis information across multiple related experiments (e.g., time-series transcriptomics experiments) to identify trends and correlations.
The CRE Browser is an integral part of our causal analysis pipeline, and has been applied to a variety of projects within Pfizer.
Reference:
1. Chindelevitch L, Ziemek D, Enayetallah A, Randhawa R, Sidders B, Brockel C, and Huang ES, Causal Reasoning on Biological Network: Interpreting Transcriptional Changes, Bioinformatics 28: 1114-1121. 2012.
Top of Page
| REGULATORY GENOMICS POSTERS |
Updated Nov 6, 2014
RG P01: Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation
Shaoke Lou1, Heung-Man Lee1, Hao Qin1, Jing-Woei Li1, Zhibo Gao2, Xin Liu2, Landon L. Chan1, Vincent K. L. Lam1, Wing-Yee So1, Ying Wang1, Si Lok1, Jun Wang2, Ronald C. W. Ma 1, Stephen Kwok-Wing Tsui1, Juliana C. N. Chan1, Ting-Fung Chan1, Kevin Y. Yip1
1The Chinese University of Hong Kong, 2Beijing Genomics Institute - Shenzhen
Background: DNA methylation is an important type of epigenetic modification involved in gene regulation. Although strong DNA methylation at promoters is widely recognized to be associated with transcriptional repression, many aspects of DNA methylation remain not fully understood, including the quantitative relationships between DNA methylation and expression levels, and the individual roles of promoter and gene body methylation.
Results: Here we present an integrated analysis of whole-genome bisulfite sequencing and RNA sequencing data from human samples and cell lines. We find that while promoter methylation inversely correlates with gene expression as generally observed, the repressive effect is clear only on genes with a very high DNA methylation level. By means of statistical modeling, we find that DNA methylation is indicative of the expression class of a gene in general, but gene body methylation is a better indicator than promoter methylation. These findings are general in that a model constructed from a sample or cell line could accurately fit the unseen data from another. We further find that promoter and gene body methylation have minimal redundancy, and either one is sufficient to signify low expression. Finally, we obtain increased modeling power by integrating histone modification data with the DNA methylation data, showing that neither type of information fully subsumes the other.
Conclusion: Our results suggest that DNA methylation outside promoters also plays critical roles in gene regulation. Future studies on gene regulatory mechanisms and disease-associated differential methylation should pay more attention to DNA methylation at gene bodies and other non-promoter regions.
(This paper has been published online by Genome Biology and is available at http://genomebiology.com/2014/15/7/408/.)
................................................................................................................
RG P02: Epigenetic and post-transcriptional crosstalk between key players of the cancer genome
Beatrice Salvatori1 , Nenggang Zhang2, Pavel Sumazin2, Andrea Califano1
1Columbia University, 2Baylor College of Medicine
Multilayer regulation of gene expression is the foundation for the evolution of complex phenotypes of higher organisms. MicroRNAs work at the post-transcriptional level, 'orchestrating' RNA expression to sustain almost every cellular process, including aberrant states such as cancer. Very compelling is the discovery that mRNAs, pseudogenes, and long noncoding RNAs compete for the binding of microRNAs, suggesting that microRNA targets can act as modulators of microRNA activity. Reports confirmed the existence of competitive forces between transcripts in multiple cellular contexts (Cesana M et al., 2011; Tay Y et al., 2014; Kumar M et al., 2014) and concomitantly in silico dynamical models were used to identify modulators of microRNAs activity (Sumazin P et al., 2011). Recently a pan-cancer study showed that competitive interactions for microRNAs might account for a substantial fraction of the 'missing genomic variability' in tumors. H. Chiu et al. (submitted 2014) identified genetic and epigenetic variants at the loci of microRNA targets as modulators of microRNA activity, leading to dysregulation of hundreds of genes, including most of the established cancer genome. We interrogated some relevant networks that have been found broadly implicated in tumorigenesis, including global epigenetic regulators. Our analyses implicated the TET family of proteins (TET1, TET2, TET3) as targets of microRNA modulation by distal genomic variants. This family of proteins was recently discovered to catalyze the conversion of 5-methyl-cytosine to 5-hydroxymethyl-cytosine, thus contributing to DNA de-methylation (Delatte B. et al., 2014). Moreover, TETs are found frequently deleted in blood tumors and are commonly down-regulated in solid cancer, including breast, lung, and pancreas cancer (H Yang et al., 2013). Our analyses suggest that their deregulation is mediated in all subtypes of breast cancer through competition for microRNA regulation. In particular, we found significant microRNA-mediated cross-talk between TET2 and the PTEN mRNAs. Our preliminary results include biochemical validation of this regulation and its functional relevance for tumor progression.
................................................................................................................
RG P03: Somatic mutations modulate ceRNA drivers of tumorigenesis
Jing He1 , Hua-Sheng Chiu2, Pavel Sumazin2, Andrea Califano1
1Columbia University, 2Baylor College of Medicine
Pan-cancer studies have shown that competitive endogenous RNA (ceRNA) networks can cooperate with chromosome instability and abnormal DNA methylation in tumors to dysregulate tumor suppressors and oncogenes. However, ceRNA cooperative association with mutations in cancer has not been studied. Integrating data from TCGA and ENCODE, we show that the cooperation between ceRNA interactions and mutations of unknown function contribute to the dysregulation of cancer genes.
We integrated ceRNA networks and mutations in an attempt to mechanistically recover missing genomic variability of cancer genes in TCGA breast cancer biopsies. Genes have missing genomic variability in a tumor dataset when their dysregulation cannot be explained through profiling of their DNA locus. Using a group lasso regression model we showed that ceRNA drivers cooperating with somatic mutations, CNV, and methylation, could account for a large fraction of the missing genomic variability of cancer genes in breast cancer. Moreover, using a greedy-forward optimization algorithm, we identified ceRNA driver mutations that could potentially drive tumorigenesis through the ceRNA mechanism. Furthermore, we showed that driver ceRNA mutations are enriched in known and predicted binding sites of transcription factors and microRNAs.
In summary, our results suggest that somatic mutations, often of unknown function, cooperate with ceRNA regulators to alter the expression of cancer genes in breast cancer tumors.
................................................................................................................
RG P04: Diverse promoter-architectures revealed by decoding heterogeneity in high-throughtput sequence data
Leelavati Narlikar1
1 National Chemical Laboratory, India
An important question in biology is how different promoter-architectures contribute to the diversity in the regulation of transcription initiation. A major step forward has been the development of technologies like CAGE/RACE that map transcription start sites (TSSs) at high resolution in a genome-wide manner. However, the subsequent step of characterizing promoters and their functions is still largely done on the basis of previously established promoter-elements like the TATA-box in eukaryotes or the -10 box in bacteria. Unfortunately, a majority of promoters and their activities cannot be explained by the presence or absence of these few elements. Motif discovery methods like MEME identify novel overrepresented elements, but these also fail here, because TSS neighborhoods are highly heterogeneous containing no overrepresented motif. For example, one set of promoters may be characterized by elements A, B, & C, another by A & D, a third only by D, and a fourth by E & F. In such a scenario, there is little chance that all six elements and four promoter-architectures will be detected by conventional approaches. Things get even more complicated when spacing between elements becomes relevant.
I will present a new unsupervised machine learning-based method designed to explicitly characterize this heterogeneity, while simultaneously unraveling underlying promoter-architectures. The method is generalizable to any organism, identifying previously undetected elements with lengths ranging from a single base in bacteria to 15 bases in certain human promoter-architectures. A striking example is the clear presence of a pyrimidine right before the TSS under very specific circumstances, across five different bacteria, which is likely to play a crucial role for transcription initiation. In tuberculosis, analysis of TSS locations across two environmental conditions provides convincing evidence that the spacing between the -10 box and the TSS is utilized for dynamic regulation of gene-expression by the pathogen. This relationship between the spacing and transcription activity has not been identified before.
In the well-studied Drosophila, the method identifies new variants of the INR motif instrumental during development, along with several novel promoter-architectures. In humans, there appears to be a lot more heterogeneity than reported so far: 20 architectures composed of a few known and many novel elements are identified, with each architecture having distinct evolutionary patterns, cell-type specific activity, and chromatin state.
The applicability of this method extends beyond identifying new promoter-architectures. This new way of looking at high-throughput sequence data allows for the identification of diverse regulatory signals associated with any DNA specified biological event reported at high-resolution.
................................................................................................................
RG P05: Computational identification of protein binding sites on RNAs using high-throughput RNA structure-probing data
Xihao Hu1 , Thomas K. F. Wong2, Zhi John Lu3, Ting-Fung Chan1, Terrence Chi-Kong Lau4, Siu-Ming Yiu2, Kevin Yip1
1The Chinese University of Hong Kong, 2The University of Hong Kong, 3Tsinghua University, 4City University of Hong Kong
High-throughput sequencing has been used to probe RNA structures, by treating RNAs with reagents that preferentially cleave or mark certain nucleotides according to their local structures, followed by sequencing of the resulting fragments. The data produced contain valuable information for studying various RNA properties. We developed methods for statistically modeling these structure-probing data and extracting structural features from them. We show that the extracted features can be used to predict RNA "zip codes" in yeast, regions bound by the She complex in asymmetric localization. The prediction accuracy was better than using raw RNA probing data or sequence features. We further demonstrate the use of the extracted features in identifying binding sites of RNA binding proteins from whole-transcriptome gPAR-CLIP data.
................................................................................................................
RG P06: Loregic: a method to characterize the cooperative logic of regulatory factors
Daifeng Wang1 , Koon-Kiu Yan1, Cristina Sisu1, Chao Cheng2, Joel Rozowsky1, William Meyerson1, Mark Gerstein 1
1Yale University, 2Dartmouth Medical School
The topology of the gene regulatory network has been extensively analyzed. Now, given the large amount of available functional genomic data, it is possible to go beyond this and systematically study regulatory circuits in terms of logic elements. To this end, we present Loregic, a novel computational method that integrates gene expression and regulatory network data, to identify and characterize the cooperativity of regulatory elements using logic-circuit models. We describe the basic regulatory triplet consisting of two regulatory factors (RFs) acting on a common target gene, using a two-input-one-output logic gate model. We use binarized gene expression data, to score the agreement between a triplet's cross-sample expression characteristics with the idealized expression pattern of each of all 16 possible logic gates (e.g., AND or XOR). A high score suggests a strong cooperativity between the regulatory activities of the two RFs following the corresponding logic gate pattern. To demonstrate Loregic's applicability, we apply it to yeast cell cycle and human cancer datasets. In yeast, we use Loregic to study yeast transcription factors (TFs) regulatory activity, and validate the results using TF knockout experimental datasets. Next, using human ENCODE ChIP-Seq and TCGA RNA-Seq expression data, we are able to demonstrate how Loregic characterizes complex circuits involving miRNAs and both proximally and distally regulating transcription factors (TFs). We find that in acute myeloid leukemia, the oncogenic TFs such as MYC, can be modeled as acting independently from other TFs, but antagonistically with miRNAs. Next, we explore the algorithm's applicability to other regulatory features. We use Loregic for the discovery and classification of indirectly bound TFs. We also predict logical operations in feed-forward loops, a special type of regulatory triplet in which one TF regulates both the target gene and the other TF. Finally, we demonstrate that Loregic is able to identify the regulatory pathways to targets that have cascaded logic-circuit operations. In summary, Loregic is a valuable computational method that describes the complex process of gene regulation in terms of the regulatory cooperative logic. The present method can be further extended to analyze cooperativity among arbitrary regulatory elements such as long non-coding RNAs and pathways. We make Loregic freely available as a general-purpose tool via https://github.com/gersteinlab/Loregic.
................................................................................................................
RG P07: The role of PIWI interacting RNAs in LINE-1 evolutionary dynamics
Leanne Whitmore1 , Debjit Ray1, Wenfeng An1, Ping Ye1
1 Washington State University
Transposons, segments of DNA that have the ability to move throughout the genome, have been active in mammalian genomes for millions of years, driving evolution by generating genetic and epigenetic changes in the genome. The autonomous retrotransposon LINE-1 (L1) uses an RNA intermediate to move and makes up approximately 17% of the human genome. L1 retrotransposition is potentially detrimental to the organism by causing disruptions in the coding sequences of genes as well as regulatory regions. Throughout evolution the sequence of L1 elements has changed, most notably the 5' untranslated region (5'UTR), which contains an internal promoter and regulates L1 transcription. The biological mechanism driving the evolution of L1 elements is not yet understood. PIWI interacting RNAs (piRNA), small non-coding RNAs that are generated from L1 transcripts, can repress L1 transcription by targeting DNA methylation to the promoter regions. Further repression of L1 activity occurs during piRNA genesis in which L1 transcripts are degraded. As the 5'UTR sequence is overrepresented in prentatal piRNAs, we hypothesize that piRNA-mediated repression plays a major role in driving L1 lineage succession. To test this hypothesis we analyze piRNA abundance toward families of mouse L1s. Our preliminary analysis shows that piRNA abundance is highest toward younger and transcriptionally more active L1 families, suggesting that the piRNA system selectively acts against active L1 families, leaving room for new families to emerge.
................................................................................................................
RG P08: Deciphering functional mechanisms for non-coding genetic variants associated with complex traits
Cynthia Kalita1 , Gregory Moyerbrailean1, Roger Pique-Regi1, Francesca Luca1
1 Wayne State University
GWAS (Genome wide association study) has identified thousands of SNPs associated with complex traits. However, generally each SNP has a small effect and it is very challenging to identify the causal one and its underlying mechanism. Many GWAS signals are in non-coding regions, so they may disrupt gene regulatory sequences such as transcription factor (TF) binding sites. Here we focus on functional annotations we previously developed by integrating binding sites predicted by a motif model with DNase I footprinting data. Using an empirical Bayesian framework implemented in the fgwas software, data from GWAS studies was combined with these functional annotations. We observed improved posterior probability of association and increased interpretability of each signal, as compared to other annotations (e.g. distance to the TSS). For lipid levels in particular, the majority of the motifs correspond to TFs involved in inflammatory pathways. In contrast, the most enriched motifs for human height correspond to TFs important for development, cell proliferation, and maintenance of stem cell state.
From the enriched motifs, we selected two with GWAS hits for LDL and total cholesterol, respectively, to validate our computational predictions using reporter gene assays. Constructs containing either the reference or alternate allele at each SNP were assayed. We show that genetic variants predicted to disrupt TF binding can drive differential gene expression. Furthermore, the direction of gene expression changes confirms genetic effects from eQTL studies (GTEX consortium).
................................................................................................................
RG P09: Improving position weight matrix-based prediction of transcription factor binding sites by integrating DNA shape features
Jichen Yang1 , Stephen Ramsey1
1Oregon State University
DNA sequence-dependent binding of transcription factors (TFs) to specific sites in the genome is central to gene regulation. Transcription factor binding site (TFBS) sequence patterns are often characterized by a position-nucleotide weight matrix (PWM) because it can be estimated from a small number of representative TFBS sequences. However, because the PWM probability model assumes independence between individual positions within the binding site, the PWMs for some TFs are poor discriminants of TFBS sequences from non-binding-site, noncoding DNA. Since three-dimensional DNA structure (i.e., the "shape" of the double helix) is recognized by TFs and is a determinant of binding specificity that depends on multi-base patterns, we investigated whether DNA shape-derived features could improve PWM-based prediction of TFBS. We defined unique features derived from base pair-level DNA shape parameters and integrated them with PWMs in a classifier for discriminating binding site sequences for 119 human TFs from random noncoding DNA sequences. We found that binding site prediction performance for 1/3 of TFs was significantly better in the shape+PWM method vs. the traditional PWM method, while the two methods had equivalent performance for the remaining TFs. Our findings indicate that DNA shape-derived features can conditionally improve PWM-based detection of TFBS, and that this improvement is in general due to the ability of DNA shape features to capture interdependencies between nucleotide positions that cannot be captured in a PWM.
................................................................................................................
RG P10: Inferring differential alternative splicing from paired-end RNA-Seq data
Ruolin Liu1 , Karin Dorman1, Julie Dickerson1
1Iowa State University
Alternative splicing (AS) is a post-transcriptional regulation mechanism under which a single gene produces multiple mRNA transcripts, called isoforms. The direct outcome of AS is to expand the diversity of mRNAs produced from the genome and, after translation, the protein diversity. Regulation of isoform abundance can have profound functional effects, including changes in protein-protein binding, protein localization, protein enzymatic properties, and protein-ligand binding, and changes in alternative splicing have been linked to human diseases. More than 95% of human genes are alternatively spliced. RNA sequencing (RNA-Seq) has emerged as a high-throughput technology capable of performing detailed transcript data surveys. As RNA-Seq becomes the standard for studying gene and isoform expression, a key problem is to detect differential alternative splicing. In this study, we propose a new method for detecting differential isoform expression using RNA-Seq. Following Rossell et. al (2014), we take a model-based approach for count data, where multiple, a priori known, isoforms contribute to each count. We extend the model to detect differential splicing, while accounting for overdispersed counts between biological replicates.
................................................................................................................
RG P11: Evaluation of the accuracy of enhancer-target associations
Qin Cao1 , Kevin Yip1
1The Chinese University of Hong Kong
Motivation: Enhancers are essential regulatory elements that play critical roles across a wide range of cellular processes. Previous studies have suggested that mutation of enhancers may lead to abnormal gene expression and result in disorders. An important step towards a complete understanding of enhancer roles is to associate the target genes regulated by each enhancer. Since enhancers can regulate gene expression via long-range interactions, experimental approaches such as Hi-C and ChIA-PET could help identify enhancer targets. However, the low resolution, high noise level and limited data availability restrict their current use in finding enhancer targets. As a result, computational approaches have been proposed as an important alternative. These methods consider activity correlations, distance information, and co-evolution signals in identifying potential targets of enhancers. The accuracy of these predictions remains to be evaluated comprehensively.
Methods and results: Here, we reason that these putative enhancer-target associations can be evaluated in silico by checking whether the activities of the involved enhancers can accurately predict the expression of the potential targets, or can significantly improve the predictions of other features. This evaluation is non-trivial because 1) a large number of features have been used in calling targets, which should not be used in the evaluation process; and 2) one enhancer can regulate multiple target genes and multiple enhancers can regulate the same gene, making the relationship between enhancer activity and target gene expression fairly complex. In view of this, we have carefully chosen enhancer features and designed data selection and cross-validation procedures that can avoid various types of bias. Based on the potential enhancer-target associations reported in several previous studies, we found that enhancer features not involved in calling their targets could indeed partially indicate the expression levels of their potential targets, although their predictive power is in general not as strong as promoter and gene body features, and the prediction accuracy depends highly on the particular data set and testing configuration. Our results also suggest that enhancers may have stronger effects on their targets in complex multiple-to-multiple enhancer-target relationships. Overall, our study provides an objective evaluation of current potential enhancer-target lists, and suggests way to improve the calling of enhancer targets.
................................................................................................................
RG P12: GWAS next generation: identifying mechanisms of action in association studies.
Maria Rodriguez Martinez1 , Paola Nicoletti2, Damien Arnol1, Andrea Califano2
1IBM, Zurich Research Laboratory, 2Columbia University
In recent years, genome wide association studies (GWAS) have identified a plethora of genetic variants associated with complex phenotypes and disease. However, many of the identified variants map to intergenic regions or lie close to genes with unknown biological connection to the disease, and thus, interpreting their functional role remains a daunting task. To tackle this problem, we have designed gVITaMIN (Genetic Variability IdenTifies Missing INteractions), an algorithm that examines the molecular mechanisms underlying the association between genetic variants and complex phenotypes. Specifically, the algorithm tests whether a genetic variant modulates the expression level of a gene or the transcriptional activity of a transcription factor, by altering the relationship with its targets.
We have applied gVITaMIN to the study of breast cancer, a common complex disease with incompletely characterized genetic predisposition architecture. We have selected 50 SNPs previously associated with breast cancer susceptibility, run gVITaMIN using two different breast cancer expression datasets (TCGA and METABRIC), and compared the results obtained from both cohorts. Interestingly, gVITaMIN links the cancer susceptibility conferred by rs1876206 to dysregulation of TGFβ signaling, a potent growth inhibitor with tumor-suppressing activity.
................................................................................................................
RG P13: Are computationally predicted footprints result of DNase I cleavage bias?
Eduardo G. Gusmao1 , Martin Zenke2, Ivan G. Costa1
1RWTH University Aachen, 2RWTH University Aachen Medical School
DNase I digestion followed by massive sequencing (DNase-seq) has proven to be a powerful technique for identifying active transcription factor (TF) binding sites on a genome-wide scale [1, 2, 3, 4]. Several computational approaches have been proposed to find nucleotide-resolution footprints, regions with 5 to 20 bps within two DNase-seq peaks [2, 4]. Recently, He et al. (2014) demonstrated that DNase-seq signal has biases reflecting the preference of DNase I to cleave particular sequences. Moreover, they show that the performance of a digital footprint method correlates with the cleavage bias of the underlying TF motif and that footprints are outperformed by simple DNase hypersensitivity sites tag count scoring (DHS-TC). However, these results were based on footprints predicted with a simple version of the digital footprint occupancy score (FOS) from [4] and no attempt was made to correct sequence bias previous to footprint prediction.
To address these questions, we extended our segmentation-based digital footprinting framework (HINT - HMM-based identification of TF footprints) [2] by performing bias correction of DNase-seq signals (HINT-BC). We estimated DNase I cleavage bias as in [3] on ENCODE DNase-seq data sets obtained from the Crawford lab (H1-hESC, HeLa-S3, Huvec and K562) and the Stamatoyannopoulous lab (HepG2, Huvec and K562). We observed that cleavage bias is distinct for each DNase-seq data set and that differences were larger between experiments from distinct labs. We then executed HINT, HINT-BC, DHS-TC, and FOS on these data sets and evaluated predictions with 139 TF ChIP-seq data sets measured on these cell types. Performance of methods were evaluated regarding their area under the ROC curve (AUC) at 10% false positive rate. Results indicate that HINT-BC significantly outperforms all compared methods, while FOS was outperformed by all methods (Friedman-Nemenyi hypothesis test at 0.05 significance level). This reinforces our point that the method evaluated in [3] is not a good representative of footprint detection methods and that footprint methods profit from sequence bias correction.
Next, we measured the correlation between observed and expected number of DNase cleavage sites around each TF. This statistics measures the potential "bias score" of a TF motif for a given DNase-seq assay [3] (Fig. 6). We observed a high negative correlation between FOS AUC and the "bias score" (-0.41, p-value < 0.00001) for all evaluated motifs, which agrees with the observation that FOS footprints are affected by DNase cleavage bias. HINT and HINT-BC presented negative correlation values of -0.14 and -0.04 (p-values > 0.05). These results show that the impact of DNase-seq cleavage bias is low on robust digital footprinting methods and can be further decreased after the correction of DNase-seq signal.
References
1. Crawford, G.E. et al. (2006). Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research, 16(1), 123-131.
2. Gusmao, E.G. et al. (2014). Detection of active transcription factor binding sites with the combination of DNase hypersensitivity and histone modifications. Bioinformatics.
3. He, H.H. et al. (2014). Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Meth, 11(1), 73-78.
4. Neph, S. et al. (2012). An expansive human regulatory lexicon encoded in transcription factor footprints. Nature, 489(7414), 83-90.
................................................................................................................
RG P14: Understanding myelome multiple patients recall by automatic reasoning on an integrated model of transcriptomic data and large-scale signaling pathways
Bertrand Miannay1 , Carito Guziolowski1, Stephane Minvielle2, Morgan Magnin1, Florence Magrangeas2
1Ecole Centrale de Nantes, 2Centre de Recherche en Cancérologie Nantes-Angers
Multiple myelome (MM) is an incurable haematological malignancy cancer; our aim is to better understand mechanisms of relapse by using expression profiles. In this work, we studied the consistency of a large-scale causal network of signaling and transcriptional events with respect to gene expression profiles from 32 patients (9 healthy, 11 measured at MM diagnosis and 12 at MM recall).
Firstly, we automatically build the regulatory network of MM cancer by connecting components of NFkappaB (RelA and NFkappaB1), a protein relevant for this cancer with differentially expressed genes across all patients. For this step we used the Pathways Interaction Database (NCI-PID). Secondly, we studied if the logic given by the causal flow events of the regulatory network is consistent with the up or down expression-shifts when comparing MM patients' expression data with respect to healthy profiles.
The logic confrontation of this data was done with a tool using a qualitative modeling approach in which each component of the system shift of expression must be explained by the shift of its predecessors, in an exhaustive manner.
Our results provide a causal and signed (activations, inhibitions) graph, composed of 961 nodes, and 1234 edges. This graph provides putative explanations for the expression-change of the variant genes in all patients. The graph causality was found much more consistent when using healthy patients data (35% inconsistency score, IS) with respect to MM patients data (45% IS). These results may imply that NCI-PID regulatory information better explains the qualitative logic of healthy cells; whereas, it presents incompleteness in describing the logic of cancer cells. Moreover, the inconsistency of expression profiles measured at diagnostic (49% IS) is slightly higher than the one measured at relapse (42% IS), suggesting that NCI-PID regulatory knowledge explains better relapse than diagnostic data. This result is supported by the fact that cancer treated cells are subject to a Darwinian selection, implying that cancer cells populations after treatment becomes more homogenous, and in one sense, some of the regulatory mechanisms are more canonical (consistent with NCI-PID).
In conclusion, this study provides a consensual measure for healthy and cancer patients, taking into account a global logical reasoning of large-scale regulatory pathways despite high variability of the patients' expression profiles. Our results can be seen as signatures of cancer stages, and in this context we consider this approach as novel and complementary to machine learning ones. We will pursue this research project by expanding the number of patients, integrating RNA-seq and ChIP-seq data in our models and link these genomic profiles to functional pathways such as apoptosis and proliferation.
................................................................................................................
WITHDRAWN
RG P16: Genetic variation and geographical implication of Moringa oleifera accessions in Nigeria using amplified fragment length polymorphism (AFLP)
Jacob Popoola1, Conrad Omonhinmin1
1Covenant University, Nigeria
Moringa oleifera is an underutilized tree crop that exists in varied geographical areas within Sub-Saharan Africa and deserves careful genetic assessment. In this study, AFLP marker was employed to evaluate the intra-specific genetic variation among 40 accessions of M. oleifera collected from different areas in Nigeria that were introduced from different countries. Six AFLP selective primer combinations were screened for their ability to generate AFLP polymorphic bands and based on the results of the banding patterns, two primer combinations (M-CAC/E-ACC and M-CAG/E-ACA) were selected. Principal coordinate analysis (PCA) and cluster analysis (CA) were employed to analyze the relationships among the 40 accessions. The primer combinations generated a total of 1272 amplification bands (primer M-CAG/E-ACA generated 859 bands while M-CAC/E-ACC generated 413 bands) out of which 1252 were polymorphic (98.43%), with size ranging from 100 to 1000 bp. High gene diversity (0.973) and polymorphic information content (0.974) were recorded for the accessions. The first two eigenvectors of PCA accounted for 18.21% of the total variation and grouped the 40 accessions into four groups. Similarity coefficient from CA ranging from 0.73 to 0.94 segregated the 40 accessions into six groups. The analysis of the clusters revealed that some accessions with similar areas of collection and background were widely separated, others clustered along collection lines. Accessions that are far apart based on their genetic similarity coefficient (KnN077, ogN026, oyN003 and edN037) could be selected for future breeding trials.
................................................................................................................
RG P15: Phenotypic and RAPD Intra-specific variability in some accessions of underutilized African yam bean (Sphenostylis stenocarpa, Hochst. ex A. Rich, Harms).
Jacob Popoola1, Mary Adebayo2, Olawale Ezekiel1, Emmanuel Adegbite3
1Covenant University, Nigeria 2International Institute of Tropical Agriculture (IITA), 3Ondo State University of Science and Technology
Intra-specific variability study was carried out on 10 accessions of African yam bean (AYB) (Sphenostylis stenocarpa, Hochst. ex A. Rich, Harms) collected from the International Institute of Tropical Agriculture (IITA) in Ibadan, Nigeria. Fourteen (14) morpho-metric characters and nine (9) arbitrary RAPD primers were used to evaluate genetic intra-specific variability among the accessions. A total of 410 bands were generated out of which 261 were polymorphic (63.66%). The significant correlation among the consistent characters such as days to 50% flowering and pods per peduncle, number of locules per pod, number of seeds per pod, pod length and seed set percentage points to their suitability for breeding and genetic improvement purposes. RAPD cluster analysis using NYSYS-pc and UPGMA program produced two major clusters with one minor cluster with similarity ranging from 72% to 93% while morph-metric clusters produced three major groups. Two accessions TSs 56 and TSs 94 had the highest level of similarity index of 93%. The use of both means had not only enabled their characterization, RAPD has eliminated selection error that may arise based on areas of collection or origin. Genetic diversity studies are very important in selection of good character traits, genetic improvement and conservation.
................................................................................................................
RG P17: STAT4 regulated transcriptional and epigenetic specification of human Th1 cells
Sini Rautio1 , Sanna Edelman2, Yuka Kanno3, Jussi Jalonen2, Subhash Tripathi2, John O'Shea3, Harri Lähdesmäki1 and Riitta Lahesmaa2
1 Aalto University, 2Turku Centre for Biotechnology, University of Turku and Åbo Akademi University, 3National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health
Signal transducer and activator of transcription 4 (STAT4) is a key factor driving the differentiation program of T helper 1 (Th1) cells. However, the genome-wide STAT4 binding and its role in controlling gene expression and epigenetic landscape have not been characterized in human Th1 cells before. By performing STAT4 siRNA silencing followed by ChIP-seq we identified genome-wide STAT4 binding sites in early differentiating and fully differentiated Th1 cells originating from naïve cord blood CD4+ Th cells activated and cultured in vitro in Th1 polarizing condition. In addition, we identified STAT4-regulated genes and global STAT4-dependent epigenetic active (H3K4me3) and repressive (H3K27me3) promoter modifications and p300 coactivator and H3K27ac recruitment to enhancers.
Preliminary results show that STAT4 binds over 25,000 loci with little changes in the binding targets over the course of differentiation. We identified genes controlled by distal or proximal STAT4 binding. STAT4 was observed to both enhance and block p300 recruitment to distal regulatory sites (enhancers), as well as activate and repress gene expression and to modulate promoter modifications of STAT4 regulated genes. STAT4 regulates many genes important in Th1 cell differentiation but also Th2 and Th17 subsets specific genes. An integrative analysis of STAT4 binding sites and immune mediated diseases associated SNPs revealed several shared loci. STAT4 binding sites were found to overlap with SNPs associated with diseases such as Crohn's disease and multiple sclerosis. In summary, the results outline the important role of STAT4 in controlling the transcription by both distal and proximal regulation and shaping the chromatin configuration, and suggests a potential role in disease etiology.
................................................................................................................
RG P18: A Bayesian multi-scale Poisson model for detecting differences in high-throughput sequencing data between multiple groups and its application to small sample sizes
Heejung Shim1 , Zhengrong Xing1, Ester Pantaleo1, Matthew Stephens1
1University of Chicago
Identification of differences between multiple groups in molecular and cellular phenotypes measured by high-throughput sequencing assays is frequently encountered in genomics applications. For example, common problems include detecting differential gene expression between multiple conditions using RNA-seq and detecting differences in transcription factor binding/chromatin accessibility across tissues using DNase-seq or ChIP-seq. Motivated by WavetQTL, our previous wavelet-based approach to genetic association analysis of functional phenotypes that better exploits high-resolution information from high-throughput sequencing assays, here we present multiseq, statistical methods that model the count nature of the sequence data directly using multi-scale models. Specifically, multiseq considers the data as an inhomogeneous Poisson process and tests for differences in underlying intensities using a Bayesian multi-scale model. We compared multiseq to WaveQTL on simulated data sets with different sample sizes or different library read depths. As expected, multiseq outperforms WaveQTL in smaller sample sizes. Even with larger sample sizes (e.g., 70), multiseq outperforms WaveQTL unless library read depths are very high. In addition, we applied those two multi-scale methods and a window-based approach, DESeq, to ATAC-seq data measured on Copper-treated samples and control samples (3 vs 3), and we found that multiseq detected substantially more differences in chromatin accessibility between two conditions than WaveQTL and DESeq.
................................................................................................................
RG P19: Progressive promoter element combinations classify conserved orthogonal plant circadian gene expression modules
Sandra Smieszek1
1Cleveland Clinic
We aimed to test the proposal that progressive combinations of multiple promoter elements acting in concert may be responsible for the full range of phases observed in plant circadian output genes. In order to allow reliable selection of informative phase grouping of genes for our purpose, intrinsic cyclic patterns of expression were identified using a novel, non-biased method for the identification of circadian genes. Our non-biased approach identified two dominant, inherent orthogonal circadian trends underlying publicly available microarray data from plants maintained in constant conditions. Furthermore, these trends were highly conserved across several plant species. Four phase-specific modules of circadian genes were generated by projection onto these trends and, in order to identify potential combinatorial promoter elements that might classify genes into these groups, we used a random forest pipeline which merged data from multiple decision trees to look for the presence of element combinations. We identified a number of regulatory motifs which aggregated into coherent clusters capable of predicting the inclusion of genes within each phase module with very high fidelity and these motif combinations changed in a consistent, progressive manner from one phase module group to the next, providing strong support for our hypothesis.
................................................................................................................
RG P20: An integrated network approach for the identification of functional large intergenic noncoding RNAs
Jiajian Zhou1, Huating Wang1, Hao Sun1
1The Chinese University of Hong Kong
Increasing evidence has indicated that large intergenic non-coding RNAs (lincRNAs) are a novel family of gene regulators. With many novel lincRNAs having been identified using high-throughput sequencing approach, computational exploration of the potential functions of novel lincRNAs will be ultra-important to prioritize and shortlist the possible novel functional lincRNAs for further studies. In this work, we used a systems biology approach to develop an integrative network by combining the regulatory data and gene expression data obtained ChIP-seq and RNA-seq together. We also developed a ranking method to evaluate the importance of key lincRNA nodes in the network. When applying this network on a public dataset on mESC, we can identify more than 70% of the reported functional lincRNAs as key functional lincRNA nodes in our network. Altogether, our approach has been demonstrated to be useful for lincRNA functional annotation.
................................................................................................................
RG P21:A discriminatory approach for transcription factor binding prediction using DNase I hypersensitivity data
Juhani Kähärä1 , Harri Lähdesmäki1
1Aalto University
Transcriptional regulation is largely controlled by DNA binding proteins called transcription factors. Understanding transcription factor binding is integral to understanding gene expression and the function of gene regulatory networks.
Currently transcription factor binding sites are determined by chromatin immunoprecipitation followed by sequencing, but this method has several limitations. To overcome these caveats, DNase I hypersensitive sites sequencing is increasingly being used for mapping gene regulatory sites. Computational tools are needed to accurately determine transcription factor binding sites from this new type of data.
In this work a novel method is developed for detecting transcription factor binding sites using DNase I hypersensitivity data. The method utilizes feature selection for choosing relevant features from DNase-seq data for optimal discrimination of bound and unbound genomic sites. The procedure is designed to ignore features resulting from the intrinsic sequence bias of the DNase I and to choose only features that improve the binding prediction performance.
The method is applied to 57 different transcription factors in cell type K562. We demonstrate that the prediction performance of the method exceeds the performance of other existing methods. Our results indicate that DNase I hypersensitivity data should be used in multiple resolutions instead of the highest possible resolution. We also show that the binding predictions should be made separately for each transcription factor and that the sequencing depth of currently available data sets is sufficient for binding predictions for most transcription factors. Finally, we show that models built with our method generalize between different cell types, making the method a powerful tool in transcription factor binding predictions using DNase I hypersensitivity data.
................................................................................................................
RG P22:Characterization of enhancer gene interactions using DNaseI and gene expression data cross 110 cell types
Pouya Kheradpour1 , Manolis Kellis1
1Massachusetts Institute of Technology
Recent efforts to characterize diseases through genome-wide association studies and annotate the genome using ChIP-seq experiments have led to a dramatic increase in putative functional genomic regions. While most of the implicated loci have fallen outside coding regions and are thought to be regulatory in nature, efforts to link these regions to their target genes, thereby permitting a better understanding of their importance, has lagged considerably. Generally, experimental linking techniques only permit the interrogation of a small number of specific regions or produce a genome-wide linking at very low resolution.
We utilize DNaseI hypersensitivity sites (DHS) and expression data from 110 human cell types produced by the ENCODE and Roadmap Epigenomics projects to produce linking confidences between hypersensitive regions and nearby genes. We find that high confidence links are supported by independent datasets such as eQTL annotations and tend to show preserved synteny across mammals.
Beyond producing these links, a careful analysis of the distribution of correlations for each gene allows us to address a number of fundamental questions in enhancer biology. We estimate the number of enhancers per gene and where they are distributed with respect to the TSS. We find bulk signal for linking to hypersensitive sites as far as 10 megabases away from a gene's TSS - substantially in excess of the distances linking or eQTLs are generally considered.
We find that linking is influenced by the presence and orientation of other nearby genes, and genomic features such as CpG islands. We also examine how these estimates and our ability to identify DHS-gene links change as we vary the number of cell types in our analysis, allowing us to extrapolate what will happen as more data becomes available.
By examining the correlation distribution correcting for distance, we are able to support the biologically established insulating role for CTCF. Applying the same methodology to all ENCODE ChIP-seq datasets and hundreds of regulatory motifs predicts other factors that are also associated with increases or decreases in enhancer linking, suggesting a more complex picture of enhancer targeting.
................................................................................................................
RG P23:Using epigenomics data to predict gene expression in lung cancer
Jeffery Li1, Travers Ching2, Sijia Huang2, Lana Garmire2
1Johns Hopkins University, 2University of Hawaii Cancer Center
Epigenetic alternations are known to be correlated with changes in gene expression. However, quantitative models that accurately predict the expression of gene expression are currently lacking. DNA methylation and histone modification are two major mechanisms of epigenetic regulation. Together, these data can accurately predict gene expression in lung cancer.
................................................................................................................
RG P24:Transcriptome profiling of pediatric core binding factor AML
Chih-Hao Hsu1 , Cu Nguyen1, Rhonda Ries2, Chunhua Yan1, Qing-Rong Chen1, Ying Hu1, Julia Kuhn2, Emma Geiduschek2,
Fabiana Ostronoff2, Derek Stirewalt2, Warren Kibbe1, Daoud Meerzaman1, Soheil Meshinchi 2
1National Institutes of Health, 2Fred Hutchinson Cancer Research Center
Acute myeloid leukemia (AML) is a hematopoietic malignancy that leads to dysregulation of critical signal transduction pathways and results in clonal expansion without complete differentiation. Although several adult AML studies have been reported, the pathogenesis of pediatric AML is still poorly understood. In this study, RNA-Seq analysis was performed in 64 pediatric patient samples to study the impact of different cytogenetic abnormalities on the transcript profiles in pediatric AML. Specifically, we focused on the comparison of samples with t(8; 21) and inv(16), referred to as the core binding factor (CBF) AML, and those with normal karyotype. In our study, the expression of all homeobox (HOX) genes in those with t(8; 21) and most of HOX genes in those with inv(16) were down-regulated compared to the samples with normal karyotype, suggesting the potential of dysregulation of HOX genes for the perturbation of normal hematopoiesis. In addition, we applied four different gene fusion detection methods, including Defuse, Tophat-Fusion, FusionMap, and Snowshoes-FTD, to identify gene fusion events in the pediatric AML samples. A total of 69 putative fusion events have been identified by at least two detection methods or by one method with a ChimerDB hit. Eight of 69 putative fusion events were found in ChimerDB and 6 of them are previously reported to be fusion events in AML. Furthermore, PIM3-SCO2 that was identified as a putative fusion event in 3 cases with Inv(16) was verified by RT-PCR and Sanger sequencing, suggesting that combination of gene fusion detection methods and ChimerDB can accurately identify fusion events. Differential splicing events were also identified between different cytogenetic cohorts, indicating the great influence of cytogenetic abnormalities on the whole transcriptome in pediatric AML. Our studies shed light on the novel cytogenetic changes in pediatric AML that might be useful to predict survival and treatment outcome.
................................................................................................................
RG P25: Unveiling the DNA binding specificities of oncoprotein c-Myc and its antagonist Mxi1 using novel high-throughput data
Ning Shen1 , John Horton1, Raluca Gordan1
1 Duke University
Transcription factors (TFs) that belong to the same structural family are known to have similar DNA binding specificities. Consequently, TF binding motifs (represented as position weight matrices, PWMs) are often indistinguishable for closely related factors. This is the case for c-Myc, one of the most frequently deregulated TFs in human cancers, and Mxi1, the closest c-Myc paralog and antagonist. Both TFs have a high binding affinity for E-box CAnnTG motifs, and available PWM models cannot differentiate between c-Myc and Mxi1 [1, 2]. Binding of oncoprotein c-Myc to the genome generally leads to gene expression amplification, whereas Mxi1 functions as a transcriptional repressor and tumor suppressor. Thus, Mxi1 appears to be an ideal antagonist for c-Myc. However, previous studies revealed that overexpression of Mxi1 retards but does not stop proliferation of c-Myc-expressing cancer cells, and in vivo TF-DNA binding data (from ChIP-seq experiments) identified many genomic targets bound by c-Myc but not Mxi1. These findings raise the question of whether the DNA binding preferences of these paralogous TFs are really identical, as inferred using consensus motifs and PWM models.
In this study we present the first quantitative, high-throughput analysis of the in vitro and in vivo DNA binding specificities of two closely related TFs, c-Myc and Mxi1. In contrast to what has been reported previously in the literature, by examining the binding specificity of c-Myc comprehensively, we identified a number of non-E-box motifs with much higher binding affinities for c-Myc than many CAnnTG E-box motifs. To better understand the DNA binding specificity of c-Myc and its antagonist Mxi1, we used the genomic-context protein-binding microarray (gcPBM) technology [3] to measure in vitro binding of the two TFs to ~50,000 putative genomic target sites, as identified by ChIP-seq in vivo. The gcPBM results show that c-Myc and Mxi1 have significant differences in their in vitro binding specificities to genomic sequences. Intriguingly, both non-canonical E-box motifs and non-E-box motifs are preferred differently by the two TFs, and these differences are not captured by PWM models. Furthermore, we show that the differences in the intrinsic binding specificities between c-Myc and Mxi1 are relevant for their differential genomic binding in the cell, and are also important for different c-Myc biological functions. Finally, computational models and DNA/protein structure analyses reveal possible mechanisms for the differential binding specificity of c-Myc and Mxi1. Our findings have important implications for the direct competition for DNA binding between c-Myc and Mxi1, and thus for the potential use of tumor suppressor Mxi1 in therapeutic approaches aimed at targeting the oncoprotein c-Myc.
References
1. Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K et al: TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic acids research 2006, 34(Database issue):D108-110.
2. Munteanu A, Gordân R: Distinguishing between genomic regions bound by paralogous transcription factors. In: Research in Computational Molecular Biology: 2013: Springer; 2013: 145-157.
3. Gordan R, Shen N, Dror I, Zhou T, Horton J, Rohs R, Bulyk ML: Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell reports 2013, 3(4):1093-1104.
................................................................................................................
RG P26:DNA methylation dynamics during somatic cell reprogramming
Giancarlo Bonora1 , Constantinos Chronis1, Marco Morselli1, Liudmilla Rubbi1, Matteo Pellegrini1, Kathrin Plath1
1University of California, Los Angeles
We used whole-genome bisulfite sequencing (WGBS) to assess global DNA methylation patterns at four different stages of reprogramming of mouse embryonic fibroblasts to an embryonic stem cell-like state, including early and late reprogramming intermediates. To better understand the dynamics of DNA methylation and how it is reset to the pluripotent state during the course of reprogramming, we compared differentially methylated regions (DMRs) with change in other genomic features, including chromatin modifications and transcription factor binding. Furthermore, we investigated the utilization of multi-restriction enzyme digestion of genomic DNA in combination with bisulfite sequencing (MRE-BS) to determine DMRs between two samples at a fraction of cost of WGBS, and found that it compared favorably to results achieved using traditional reduced representation bisulfite sequencing (RRBS). The results of this research will shed light on epigenetic regulation during the reprogramming process.
................................................................................................................
RG P27:Hidden Markov model analysis reveals complex binding modes for the transcription factor Gcn4
Todd Riley1, Cory Colaneri1
1University of Massachusetts Boston
Many new high-throughput technologies have been developed to quantitatively measure both in vivo and in vitro protein-DNA binding. Likewise, many computational algorithms have been developed to determine the binding preferences of these transcription factors (TFs) from the binding data. Most protein-DNA specificity inference methods suffer from a major weakness in that they can model only fixed-length motifs. This is especially problematic since recent studies have revealed that many families of TFs that bind primarily as dimers and tetramers have variable-length sequences (called spacers) separating the two sequence-specific half sites that, all together, delineate the binding motif. Other recent studies have shown examples of proteins that can interface with the DNA via different regions of the protein. These different "binding modes" also introduce variability in the lengths of their respective binding sites. Additionally, some proteins exhibit degenerate DNA specificity and exhibit tolerated insertions or deletions of nucleotides in multiple places within some of their binding sites as compared to their canonical consensus motifs - which again introduces binding site length variability.
With the impetus to correctly model variable-length motifs, many researchers have used hidden Markov models (HMMs), and their generalizations, as a probabilistic framework to capture insertions or deletions (indels) of nucleotides within a binding site. The so-called profile hidden Markov model (pHMM) has a topology that is well-suited for properly modeling observed indels within a sequence and has been successfully applied to many domains. PHMM methods have recently been developed to model variable-length spacers in protein-DNA binding sites. However, there exist many possible HMM topologies for modeling variable-length spacers and each topology has different modeling characteristics. It has not been determined which HMM topology is best to capture these spacer dynamics for any given protein.
In order to identify which among the many possible HMM topologies is optimal to model a variable-length spacer within a protein-DNA binding site, we chose to analyze both protein binding microarray (PBM) and "high-throughput sequencing"-"fluorescent ligand interaction profiling" (HiTS-FLIP) binding data for the bZIP protein Gcn4. The Gcn4 protein is ideal for our study since: (1) it is known to contain a variable-length spacer, and (2) the very deep sequencing of the HiTS-FLIP binding data for Gcn4 allows for the inference of very highly accurate affinity models for analysis.
Our analysis reveals complex dependencies between the variable-length spacer and the surrounding half-sites. Furthermore, we show that the simple HMM topologies currently in use to model variable-length spacers in protein-DNA binding sites are not adequate to capture these dependencies. We propose spacer-dependent HMM models that more accurately capture the complex dependencies between the spacers and their surrounding half sites with fewer model parameters as compared to common pHMM topologies. While modeling the two known spacer-lengths of Gcn4, our spacer-dependent HMM with 58 parameters explains 80% of the variance for all measured 12-mer relative affinities of at least 0.01 (1.0 being the maximum). In addition, we show that the typical pHMM topologies currently in use contain too many parameters (≥ 84), fail to correctly capture the spacer dependencies, and generate protein-DNA affinity models with inflated nucleotide insertion and deletion rates as compared to the consensus motif.
................................................................................................................
RG P28: GREAT: Genome REgulatory and Architecture Tools. The GREAT:SCAN software suite
Costas Bouyioukos1, Mohamed Elati1, François Képès1
1Institute of Systems & Synthetic Biology, Université d'Evry
Modern advances in genomics, transcriptomics, and genome structural biology have revealed significant insights on the interdependence between genome expression and layout. Evidence for non-random genome layout [1], defined as relative positioning of co-regulated or co-functional genes, stems from two main insights. Firstly, the analysis of contiguous genome segments across species has highlighted the conservation of gene order along chromosome regions. Secondly, the study of long-range regularities within chromosomes in a given species has emphasized periodic positioning of genes that are co-regulated, co-expressed, evolutionarily correlated, or highly codon-biased [3],[5]. Tools to detect, visualize, systematically analyze, integrate, and exploit gene position regularities along genomes have been developed [2].
Here we present a software suite designed to perform a systematic and integrated analysis of regular patterns along genomes. The suite is based on an algorithm to detect periodicities and it provides an easy-to-use interface to perform complete analyses of regular patterns and to visualize results.
The suite comprises three tools. GREAT:SCAN:patterns, a package for systematic study of periodic patterns; GREAT:SCAN:integrate, a novel computational process which integrates regularities along multiple transcription factors (TFs) and chromosomes; and GREAT:SCAN:presicion, a machine learning tool to predict novel TFBS.
GREAT:SCAN:patterns performs a complete analysis of periodic patterns in three steps. The first step calculates an exact p-value for all predicted periods and returns a rank of them and a periodogram. In the second step a clustering algorithm detects clusters of genes that are "in-phase" on the modulo period coordinates, providing evidence of possible local spatial proximity of genes. In the last step a variable size sliding window performs a more fine-tuned search for regularities on specific domains of the chromosome. In this work, we present a complete analysis of 7 major TFs of E. coli and report preliminary results that regions of periodic arrangement are associated with the macro-domain organization of this bacterial genome.
GREAT:SCAN:integrate: is a computational process which automatically consolidates and integrates analysis of periodic patterns on multiple TFs and/or on multiple chromosomes. It consists of a series of seven steps. Initially, periods are detected on all the groups of co-regulated genes and then a couple of integration steps on the TF and the chromosome level consolidate periods and extend overlapping extremities. Finally, the process is searching for integer multiple periods and collects them all to form families of harmonics with their periodic extremities extended. The result of the final step is visualized as a set of periodic regions that span chromosomes and the results of each intermediate step are stored in a database for further analysis and/or visualization.
We will present the formal description of the 7 step process together with initial evidence of an application on the yeast Saccharomyces cerevisiae TF network. Where we identify common periods, harmonics and significant degree of overlap between the master transcription regulators of yeast.
The two tools are developed to detect periods on co-regulated genes; however, it can work with any gene set of interest as well as with any set of genomic positions of interest, including but not limited to ChIP-seq data.
GREAT:SCAN:precision is a novel implementation of a machine learning tool for TFBS prediction [4] which incorporates two inputs in a classifier: a) direct DNA sequence motif readout, and b) genome layout readout from the genomic coordinate. The underlying rationale is based on the emerging observation that co-regulated genes are positioned at periodic intervals along the chromosome. The combined classifier is then obtained with an iterative weight update scheme, using a modified version of the AdaBoost algorithm. We will report on the novel prediction of E. coli TFBS as well as insights on the interplay between sequence motif and position.
References
1. Képès, F. (2004). Periodic transcriptional organization of the E. coli genome. J Mol Biol 340, 957-964.
2. Junier, I., Hérisson, J., and Képès, F. (2010). Efficient detection of periodic patterns within small datasets. Algorithms for Molecular Biology 5, 31.
3. Junier, I., Hérisson, J. and Képès, F. (2012). Genomic Organization of Evolutionarily Correlated Genes in Bacteria: Limits and Strategies. J. Mol. Biol. 419, 369-86.
4. Elati, M., Nicolle, R., Junier, I., Fernandez, D., Fekih, R., Font, J. and Képès, F. (2013). PreCisIon: PREdiction of CIS-regulatory elements improved by gene's positION. Nucleic Acids Res. 41, 1406-15.
5. Képès, F., Jester, B.C., Lepage, T., Rafiei, N., Rosu, B. and Junier, I. (2012). The layout of a bacterial genome. FEBS Lett. 586, 2043-2048.
................................................................................................................
RG P29: A biophysical analysis of transcription factor binding data
Rahul Siddharthan1
1The Institute of Mathematical Sciences
In 2003, Djordjevic, Shraiman, and Sengupta proposed a biophysical approach to the binding of transcription factors, pointing out that rather than the simple statistical description offered by a PWM, a more accurate expression for the probability of a TF binding to a sequence is given by the Boltzmann factor for the binding energy E, exp(-beta (E-mu)) (where beta is the inverse temperature and mu is the chemical potential), divided by the partition function Z. In the simple case that the two options are binding or not-binding, Z = 1+exp(-beta(E-mu)), and the overall probability takes the form of a sigmoidal or Fermi function. Even if we assume that E is additive across nucleotides, this function is not multiplicative, and the PWM description of the probability as a product over individual nucleotides breaks down.
Transcription factor binding sites (TFBS) rarely occur individually and usually several are bound to a single sequence. This raises the possibility that what is important is not the "strength'' (binding affinity or probability, PWM score, etc.) for a single site, but the expected number of bound factors across a promoter or enhancer region. The thermodynamic probability of this, too, can be calculated efficiently, taking into account competition between factors for binding sites.
The difficulty, of course, is that biophysical binding energies are available for very few transcription factors, and for very few bound sequences per factor, and vary according to cell type and environmental conditions. Our approach is not to depend on experimental data on binding energy, but to use it as a description of binding whose parameters are to be inferred from experimental data on bound sequences.
Specifically, we use this idea to distinguish between bound and unbound sequences in ChIP-seq data in yeast, from Venters and Pugh. For each of 32 transcription factors for which adequate numbers of bound sequences were available, we divided the bound sequences into training and testing sequences. For each training sequence we constructed a synthetic "negative'' sequence from a Markov model constructed from the set of training sequences. We then inferred energy matrices that maximized the difference in binding between training and negative sequences.
For the testing sequences, we constructed two negative sets, each of the same size as the training data: one synthetic as above, and one of actual intergenic sequence in yeast that has not been observed to be bound by the concerned factor. We then calculated the difference in the biophysically calculated number of bound factors among these three sets. We do the same exercise with literature PWMs.
The results, though preliminary, are highly encouraging. In 20 of 32 cases, the biophysical method clearly distinguishes the bound testing regions from synthetic data. In 12 out of 32 cases, the biophysical method also clearly distinguishes the known-bound testing regions from other intergenic regions. The PWM-based approach, with its well-known propensity for false positives, also gives a higher score to the bound testing regions in many cases when literature PWMs are used, but much less significantly.
While there have been several previous attempts to go beyond position weight matrices for TFBS description, including this author's, the biophysical approach holds appeal for its physical intuitiveness and ease of calculation.
This is work in progress. Further refinement of the method and validation on other datasets, including ENCODE chip-seq data, is in progress.
................................................................................................................
RG P30: Transcription factor cooperatively reveals distinguishing characteristics of the HepG2 cell-line
Konnor La1 , Parsa Hosseini1, Yupeng Wang1, Ivan Ovcharenko1
1 National Institutes of Health
Transcription regulation is a tightly controlled biological process. One particular aspect of transcription regulation includes transcription factors (TFs) binding to specific DNA sequences known as cis-elements. With advances in high-throughput sequencing technologies, researchers can now quantify TF interplay and improve the high-resolution map of the genomic regulatory landscape.
Enhancers are one class of regulatory elements that are generally found kilobases away from or sometimes within the target gene and possess the ability to regulate transcription. Until lately, genomic repositories of such elements were largely lacking, and initiatives such as the ENCODE Consortium aspire to quantify their systematic interplay.
In this study, building upon the ENCODE's human liver carcinoma cell-line (HepG2), we quantify the HepG2 TF-TF spatial landscape. We identified numerous TFs pairs involved in liver cellular respiration. We used statistical models to quantify enrichment of TF binding sites across HepG2 enhancers and found 0.05% of possible transcription factors combinations to be significant pairs. TFs such as FOX, HNF4, HNF3, and NR1H2-RXRA, are enriched both in a spatial and sequential manner. Interestingly, when we compared HepG2-specific TF-TF pairs to eight other ENCODE cell-lines, we found that many HepG2 TF pairs were mutually exclusive to HepG2. Lastly, we show that many HepG2-specific TF-TF pairs exhibit increased conservation when compared to pairs of other cell-lines. Such results could therefore shed light on a liver-specific set of TFs that potentially function collectively to govern liver specificity.
In summary, this study examines the TF-TF spatial landscape across HepG2. Our results reveal a set of liver-specific TFs that are enriched in crucial biological processes and exhibit conservation across placental mammals.
................................................................................................................
RG P31: An evolutionarily biased distribution of miRNA sites toward regulatory genes with high promoter-driven intrinsic transcriptional noise
Hossein Zare1 , Arkady Khodursky2, Vittorio Sartorelli1
1National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), National Institute of Health (NIH), 2University of Minnesota
miRNAs are a major class of regulators of gene expression in metazoans. By targeting cognate mRNAs, miRNAs are involved in regulating most, if not all, biological processes in different cell and tissue types. To better understand how this regulatory potential is allocated among different target gene sets, we carried out a detailed and systematic analysis of miRNA target sites distribution in the mouse genome.
We used predicted conserved and non-conserved sites for 779 miRNAs in 3' UTR of 18440 genes downloaded from TargetScan website. Our analysis reveals that 3' UTRs of genes encoding regulatory proteins harbor significantly greater number of miRNA sites than those of non-regulatory, housekeeping and structural, genes. Analysis of miRNA sites for orthologous 3'UTR's in 10 other species indicates that the regulatory genes were maintaining or accruing miRNA sites while non-regulatory genes gradually shed them in the course of evolution. Furthermore, we observed that 3' UTR of genes with higher gene expression variability driven by their promoter sequence content are targeted by many more distinct miRNAs compared to genes with low transcriptional noise.
Based on our results we envision a model, which we dubbed "selective inclusion," whereby non-regulatory genes with low transcription noise and stable expression profile lost their sites, while regulatory genes which endure higher transcription noise retained and gained new sites. This adaptation is consistent with the requirements that regulatory genes need to be tightly controlled in order to have precise and optimum protein level to properly function.
................................................................................................................
RG P32: Consensus strategy improves microRNA prediction
Bin Xue1
1University of South Florida
microRNAs are short regulatory RNA with about 22 nucleotides. microRNAs are produced through at least two pathways. In the first pathway, primary microRNAs with multiple stem-loop structures are processed by microprocessor Drosha to produce precursor microRNAs, which have ~80 nucleotides forming stem-loop structure. The precursor microRNAs will be further processed by Dicer to produce mature microRNAs. In the second pathway, short RNA transcripts that have step-loop structures will be processed by Dicer directly to produce mature microRNAs. In both pathways, specific sequential features on the RNA transcripts were observed. Based on these observations, many computational strategies have been proposed to predict microRNAs based on secondary structure, base pairing free energy, or sequence conservation. These predictors are normally developed using hundreds of known microRNAs. Although very successful, the application of these predictors is still limited due to their high false positive rate. We proposed a consensus strategy to integrate the strength of different predictors and tested the performance in a much larger dataset of nearly 2000 microRNAs. Our result shows that the consensus strategy improves the prediction performance consistently. This strategy may have broad application in the future due to its remarkably reduced false positive rate.
................................................................................................................
RG P33: Post-transcriptional regulation mediated by the interplay between RNA-binding proteins and miRNAs
Atefeh Lafzi1, Saber Hafezqorani1, Yesim Aydin Son1, Hilal Kazan1
1Middle East Technical University, Turkey
Post-transcriptional regulation (PTR) is mediated by the interactions RNA-binding proteins (RBPs) and microRNAs (miRNAs) with cis-regulatory sites in mRNAs. Recent studies have found that each factor binds to hundreds of targets, and each mRNA is occupied by several factors. Also, RBPs and miRNAs are shown to function in coordination with each other in many cases [1]. However, a majority of previous research has focused on the regulatory effect of individual factors. In this study, we leveraged the recent explosion of PTR-related data to map both RBP and miRNA sites on mRNAs and considered the effect of multiple factors at the same time. We mapped RBP sites by taking into account motifs identified with RNAcompete [2], peaks from gPARCLIP and CLIP data [3-4] and PhastCons conservation scores. To map miRNA sites, we combined PicTar and TargetScan predictions, Ago2 CLIP-identified peaks [4] and experimentally supported targets from miRTarBase database [6]. We then analyzed the mapped sites concurrently to detect potential interactions. These interactions could be competitive when there is overlap between the sites or cooperative when sites of two factors are located on each side of a stem (e.g. Pum and miR-221 [1]). For HuR and IGF2BP2, we showed that mRNAs with sites which are in competition with other factors' sites show a distinct stability profile compared to mRNAs with other sites. We also studied the effect of cooperative interactions of HuR with other factors upon HuR knockdown. Lastly, we explained the distinct activities of identical PUM1 sites [7] by considering the accessibility and cooperative / competitive interactions of these sites. These results show that modeling the effect of multiple factors and their interactions concurrently improves our understanding of PTR.
References
1. Kouwenhove MV, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer 9 (2011), 644-656.
2. Ray D, Kazan H,et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499 (2013), 172-177.
3. Baltz AG, Munschauer M et al. The mRNA-bound proteome and its global occupancy. Mol Cell (2012) 46(5):674-690.
4. Anders G, Mackowiak SD et al. doriNA: a database of RNA interactions in post-transcriptional regulation. NAR (2012) D180-D186.
5. Cook KB, Kazan H et al. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res (2011) D301-308.
6. Hsu SD, Lin FM et al. miRTarBase: a database curates experimentally validated miRNA-target interactions. NAR (2011) D163-169.
7. Zhao W, Pollack JL et al. Massively parallel functional annotation of 3'UTRs. Nat Biotechnology (2014) 32(4):387-391.
................................................................................................................
RG P34: Transcriptome-wide identification of cancer-specific splicing events across multiple tumors
Yihsuan S. Tsai1 , Daniel Dominguez1, Shawn M. Gomez1, Zefeng Wang1
1University of North Carolina at Chapel Hill
Dysregulation of alternative splicing (AS) is one of molecular hallmarks of cancer, with splicing alteration of numerous genes in cancer patients. However, identification of cancer-specific AS events is complicated by large noise of tissues-specific splicing that hinders the mechanistic understanding of splicing dysregulation. To determine a signature of cancer-specific splicing, we explored large-scale transcriptome sequencing data from the TCGA project to identify a core set of cancer-specific AS events that are significantly altered across multiple cancer types. Further analyses suggest that these cancer-specific AS events are (1) altered in cancer among different tissue types; (2) highly conserved among vertebrates; (3) more likely to maintain protein reading frame than control AS events; (4) have functions related to cell cycle and cell adhesion; (5) able to serve as new molecular biomarkers of cancer. Finally we identified genes whose expression is closely associated with cancer-specific splicing, and discovered that most of these genes are key regulators of the cell cycle. This suggests that the activity of splicing factors may be controlled in a cell cycle dependent manner and thus cell cycle proteins can indirectly affect splicing in tumor cells. In summary, our work identifies a common set of cancer-specific AS events dysregulated across different types of cancers and provides mechanistic insight into how splicing might be mis-regulated in cancers.
................................................................................................................
RG P35: Integrative and personalized QSAR analysis in cancer by kernelized Bayesian matrix factorization
Muhammad Ammad-Ud-Din1, 2 , Elisabeth Georgii1,2, Mehmet Gönen1,2, Tuomo Laitinen3, Olli Kallioniemi4,5, Krister Wennerberg 4,5, Antti Poso3,6, Samuel Kaski1,2,5
1Helsinki Institute for Information Technology, 2Aalto University, 3University of Eastern Finland, 4Institute for Molecular Medicine Finland (FIMM), 5University of Helsinki, 6University Hospital Tübingen
We develop in silico models to find drugs with a potential for cancer treatment. Recent large-scale drug sensitivity measurement campaigns offer the opportunity to build and test models that predict responses for more than one hundred anti-cancer drugs against several human cancer cell lines. So far, these data have been used for searching dependencies between genomic features and drug responses, addressing the personalized medicine task of predicting sensitivity of a new cell line to an a priori fixed set of drugs. On the other hand, traditional quantitative structure-activity relationship (QSAR) approaches investigate small molecules in search of structural properties predictive of the biological activity of these molecules, against a single cell line. We extend this line of research in two directions: (1) an integrative QSAR approach, predicting the responses to new drugs for a panel of multiple known cancer cell lines simultaneously, and (2) a personalized QSAR approach, predicting the responses to new drugs for new cancer cell lines. To solve the modeling task, we apply a novel kernelized Bayesian matrix factorization method. For maximum applicability and predictive performance, the method optionally utilizes multiple side-information sources such as genomic features of cell lines and target information of drugs, in addition to chemical drug descriptors. In a case study on 116 anti-cancer drugs and 650 cell lines from Sanger Institute Wellcome Trust, we studied the usefulness of the method in several relevant prediction scenarios, differing in the amount of available information, and analyzed the importance of various types of drug features for the response prediction. We showed that the use of multiple side information sources for both drugs and cell lines simultaneously improved the prediction performance. In particular, combining chemical and structural drug properties, target information, and genomic features yielded more powerful drug response predictions than drug descriptors or targets alone. Furthermore, the method achieved high performance (RMSE=0.46, R2=0.78, Rp=0.89) in predicting missing drug responses, allowing us to reconstruct a global map of drug responses, which is then explored to assess treatment potential and treatment range of therapeutically interesting anti-cancer drugs.
................................................................................................................
RG P36: MicroRNA portal server for deep sequencing, expression profiling and mRNA targeting
Byungwook Lee1
1Korean Bioinformation Center
In the field of microRNA (miRNA) research, biogenesis and molecular function are two key subjects. Next-generation sequencing has become the principal technique for cataloging miRNA repertoire and generating expression profiles in an unbiased manner. We developed a web-based database server that compiled the deep sequencing miRNA data available in public and implemented several novel tools to facilitate exploration of massive data. The miR-seq browser supports users to examine short read alignment with the secondary structure and read count information available in concurrent windows. Features such as sequence editing, sorting, ordering, import, and export of user data would be of great utility for studying iso-miRs, miRNA editing, and modifications. miRNA-target relation is essential for understanding miRNA function. Coexpression analysis of miRNA and target mRNAs is visualized in the heat-map and network views where users can investigate the inverse correlation of gene expression and target relations, compiled from various databases of predicted and validated targets. By keeping datasets and analytic tools up-to-date, miRGator should continue to serve as an integrated resource for biogenesis and functional investigation of miRNAs.
................................................................................................................
RG P37: Detection of a fusion gene using soft-clipping reads in exome-sequencing data
Nam Jin Gu1 , Ji Woong Kim2, Ryan W Kim1
1Korean Bioinformation Center, 2UT Southwestern Medical Center
A gene fusion plays an important role in oncogenes that drive tumor formation and progression because it can produce active abnormal protein. To use next generation sequencing (NGS) technologies, a different fusion gene has been discovered in human cancers. Many computational methods are developed by RNA-Seq data, and a few WGS-Seq data for fusion gene discovery, but Exome-Seq data has not yet seen any use. We developed the new algorithm for detection of the fusion gene to use Exome-Seq data. In this approach, we first found candidate of fusion region to use soft-clipping reads, and the split reads are used for mapping fusion partner. Finally, the predicted fusion sequence was estimated for the frequency in read alignment. It is impossible to detect a fusion of introns with Exome-seq data that are far away from exon boundary. We were able to predict fusion boundary within exon that was caused by structural variations of the genome such as chromosomal translocation, deletion, and inversion by aligning the sequence to the pseudo-reference from fusion sequence. In addition, the allele frequency information of fusions could be very useful to filter them and discover the biology from the genomic aberration, especially fusion gene in samples with heterogeneity like tumor tissue.
................................................................................................................
RG P38: Topographical mapping of temporal gene activation
Daniel Morris1
1Loma Linda University
Despite limitations that prevent Pol II ChIP from rigorous quantitative application, the techniques ability to instantaneously measure of Pol II density at any position along a gene provides unique transcriptional information. Analysis of acute IEG (immediate early gene) activation following α1a Adrenergic Receptor stimulation identified rate-limiting transcriptional events that control both the speed of mRNA maturation and the extent of transcriptional upregulation. ChIP results were validated by comparison to major transcriptional events assessable by microarray and PCR analysis of precursor and mature mRNA. My data shows that initial transcriptional velocity on newly activated mammalian IEGs can be very high and approach maximal transcriptional rates. Despite the limited gene set, recently described mechanisms of co-transcriptional gene regulation were identified, including abrogation of promoter proximal pausing, internal transcriptional blockade, and polyadenylation associated pre-mRNA degradation. Importantly, although co-transcriptional regulatory mechanisms were present for most genes, increased recruitment of RNA Pol II was a substantial factor contributing to increased mRNA levels for all genes. As an example, regulation of Nr4a3 involved increased recruitment, delayed abrogation of a strong proximal pause, transcription of short and long isoforms, and an apparent decrease in transcriptional velocity for the long isform due to the internal polyadenylation site. Significantly, delayed abrogation of promoter proximal pausing implies this mechanism functions to delay transcription, probably as a means of ensuring transcriptional fidelity.
Given the generality of multilevel gene regulation, integrated analysis made possible by Pol II ChIP appears necessary to distinguish causative from potential rate limiting mechanisms. Further, the temporal and sometimes transient nature of cotranscriptional mechanisms provides important caveats to nontemporal methods. For example, recent omic analysis of transcriptional velocity used only two time points and would not have detected reduced velocity due to transient pausing within genes. In addition, nontemporal approaches could not identify the delay induced by promoter proximal pausing. Our data suggests temporal omic approaches that can address deficiencies in non-temporal analysis.
................................................................................................................
RG P39: Consensus approach to identify consistent brain gene expression signatures for neurodegenerative diseases
Raymond Yan1, Jie Quan2, Li Xi2, Simon Xi2
1Boston University, 2Pfizer
A large number of neurodegenerative disease-related gene-expression datasets have been deposited in public repositories over the years. These datasets have tremendous value for integrative data mining to uncover dysregulated biological pathways in diseased brains. However, the limited sample sizes, differences in brain collection procedures, sample characteristics, and other hidden confounding factors often decrease the confidence in changes observed in individual studies. Here, we carefully curated and reprocessed dozens of independent postmortem brain gene expression datasets of Alzheimer's and Parkinson's diseases from GEO and ArrayExpress. We used a consensus scoring scheme to identify hundreds of genes that are most consistently differentially expressed under disease conditions across studies. Many of these gene expression changes were also observed in mouse models of disease. They provided novel insights to AD- and PD-related pathways and biological processes. Comparisons of these AD and PD signature genes with genes located in GWAS risk loci further suggest potential causal genes in these regions.
................................................................................................................
RG P40: Identification of stage specific functional regulatory elements in Brugia malayi for Lymphatic Filariasis (LF) disease intervention
Rami Al-Ouran1, Elodie Ghedin2, Lonnie Welch1
1Ohio University, 2New York University
Gene transcription initiation and gene regulation are complex biological mechanisms that involve several molecular components working in a precise manner. Transcription factors (TFs) and the transcription factor binding sites (TFBSs) are functional elements that control gene regulation. Identifying TFBSs will assist in deciphering regulation of transcription and represent potential target sites for disease prevention. Lymphatic filariasis (LF), also known as elephantiasis, is a neglected tropical disease that affects over 120 million people worldwide. Brugia malayi is one of the nematode (roundworm) parasites that cause LF. Each stage of the B. malayi lifecycle has a unique gene transcriptional signature and third-stage filarial larvae (L3) are of particular interest as they represent the infective stage. In this study we aim to discover the putative TFBSs that are unique to genes over expressed in the L3 phase of the B. malayi lifecycle. Identifying the B. malayi stage specific regulatory elements could help in developing intervention strategies for the control of LF.
................................................................................................................
RG P41: Design principles of circadian systems
Nandita Damaraju1 , Karthik Raman2
1Georgia Institute of Technology, 2Indian Institute of Technology Madras
Circadian rhythms are biological processes, which have time periods of approximately 24 hours. Circadian networks orchestrate a variety of processes in a diverse set of organisms as simple cyanobacteria and more complex organisms such as mammals. This raises an interesting question of whether such diverse organisms have common design principles underlying their circadian networks. Previous attempts have derived design principles by studying the preexisting circadian regulatory networks in organisms. Such approaches do not exhaustively search for motifs that could potentially give rise to more robust and sustained oscillations. In this study, a thorough unbiased search is performed across all possible topologies, to identify motifs that give rise to circadian oscillations. To identify such features, all two-node and three-node networks were enumerated and their interactions were dynamically modeled. Only a few networks capable of producing oscillations were observed. These favorable topologies were then analyzed to identify common motifs. The motifs obtained were consistent with the existing circadian networks of organisms, thereby successfully identifying the core features responsible for circadian oscillations. This study identifies the design principles of circadian networks in an unbiased fashion and answers questions about the minimum requirements to achieve circadian oscillations while highlighting the key topological and dynamical features of such networks. The results obtained could be used to gain valuable insights into the circadian mechanisms across varied organisms and could help potentially build more complex systems with and custom targeted behaviors.
................................................................................................................
RG P42: A pooling-based approach to mapping genetic variants associated with DNA methylation
Irene Kaplow1 , Sarah Mah2, Julia MacIsaac2, Michael Kobor2, Hunter Fraser1
1Stanford University, 2University of British Columbia
DNA methylation is an epigenetic modification that plays a key role in gene regulation. Previous studies have investigated its genetic basis by mapping genetic variants that are associated with DNA methylation at specific sites, but these have been limited to microarrays that cover less than 2% of the genome and cannot account for allele-specific methylation (ASM). Other studies have performed whole-genome bisulfite sequencing on a few individuals, but these lack statistical power to identify variants associated with methylation. We present a novel approach in which bisulfite-treated DNA from many individuals is sequenced together in a single pool, resulting in a truly genome-wide map of DNA methylation. Compared to methods that do not account for ASM, our approach increases statistical power to detect associations while sharply reducing cost, effort, and experimental variability. As a proof of concept, we generated deep sequencing data from the pooled DNA of 60 human cell lines and identified over 2000 genetic variants associated with DNA methylation. We found that these variants are enriched in tissue-specific transcription factor binding sites and can also be associated with chromatin accessibility and gene expression. In sum, our approach allows genome-wide mapping of genetic variants associated with DNA methylation in any species, without the need for individual-level genotype or methylation data.
................................................................................................................
RG P43: Integrative analysis of haplotype-resolved epigenomes across human tissues
Inkyung Jung1 , Danny Leung1, Nisha Rajagopal1, Bing Ren1
1Ludwig Institute of Cancer Research
Allelic differences between the two sets of chromosomes can affect the propensity of inheritance in humans; however, the extent of such differences in the human genome has yet to be fully explored. Here, for the first time, we delineate allelic chromatin modifications and transcriptomes amongst a broad set of human tissues, enabled by a chromosome-spanning haplotype reconstruction strategy. The resulting masses of haplotype-resolved epigenomic maps are the first of its kind and reveal extensive allelic biases in the transcription of human genes, which appear to be primarily driven by genetic variations. Furthermore, allelic resolution of chromatin states allows us to discover cis-regulatory relationships between genes and their control sequences. These maps also uncover intriguing characteristics of cis-regulatory elements and tissue-restricted activities of repetitive elements. The rich datasets described here will enhance our understanding of the mechanisms controlling tissue-specific gene expression programs.
................................................................................................................
RG P44: Comparison of D. melanogaster and C. elegans developmental stages, tissues, and cells by modENCODE RNA-Seq data
Jingyi (Jessica) Li1 , Haiyan Huang2, Peter J. Bickel2, Steven Brenner2
1University of California, Los Angeles, 2University of California, Berkeley
We report a statistical study to discover transcriptome similarity of developmental stages from D. melanogaster and C. elegans using modENCODE RNA-seq data. We focus on "stage-associated genes" that capture specific transcriptional activities in each stage and use them to map pairwise stages within and between the two species by a hypergeometric test. Within each species, temporally adjacent stages exhibit high transcriptome similarity, as expected. Additionally, fly female adults and worm adults are mapped with fly and worm embryos, respectively, due to maternal gene expression. Between fly and worm, an unexpected strong collinearity is observed in the time course from early embryos to late larvae. Moreover, a second parallel pattern is found between fly prepupae through adults and worm late embryos through adults, consistent with the second large wave of cell proliferation and differentiation in the fly life cycle. The results indicate a partially duplicated developmental program in fly. Our results constitute the first comprehensive comparison between D. melanogaster and C. elegans developmental time courses and provide new insights into similarities in their development. We use an analogous approach to compare tissues and cells from fly and worm. Findings include strong transcriptome similarity of fly cell lines, clustering of fly adult tissues by origin regardless of sex and age, and clustering of worm tissues and dissected cells by developmental stage. Gene ontology analysis supports our results and gives a detailed functional annotation of different stages, tissues, and cells. Finally, we show that standard correlation analyses could not effectively detect the mappings found by our method.
................................................................................................................
RG P45: Enhancer RNAs reveal widespread chromatin reorganization in prostate cancer cell lines
Ville Kytölä1 , Annika Kovakka1
1University of Tampere
Chromatin conformation determines the gene regulatory programs and enables the diversity of cell types. Characterization of chromatin state across different cell lines has been a central focus of major projects such as ENCODE. These studies have revealed a number of insights into cellular programs in cell differentiation and disease related dysregulation. However, the degree of chromatin variation between individuals is less studied and the diversity of chromatin organization in cancer is not known.
In order to gain insight into diversity of chromatin organization in prostate cancer we characterized 11 prostate and prostate cancer cell lines under different culture conditions using Global Run-On sequencing (GRO-seq). This assay allowed us to identify the active enhancer areas from each cell line through detection of nascent transcription of enhancer RNA (eRNA) molecules. To this end, we developed a new computational algorithm to identify eRNA signals in a genome-wide manner by utilizing the unique bi-directional pattern of nascent transcription. Identified eRNA sites show high consistency with areas of open chromatin from DNase I sequencing (DNase-seq) data as over 80% of the sites are covered by open chromatin signals in LNCaP cells.
We present a comparison of eRNA signals across prostate cancer cell lines. Our analysis reveals extensive variation in enhancer activity between prostate cancer models. On average, approximately 3000 active eRNA loci were identified from each cell line with the number of detected sites varying from 1300 to 8000. Based on the detection results, the cell lines clustered according to androgen receptor (AR) status. When cultured in the presence of androgens, the number of identified eRNA sites in LNCaP and VCaP cells doubled in comparison to cells cultured without androgens. Overall, we identified nearly 25,000 distinct loci of which only 33% were shared between more than two cell lines. We find a high number of loci for which eRNA activity correlates with the expression of nearby genes. Interestingly, from among these sites we were able to extract a subset of over a hundred extremely highly correlating ( > 0.9) connections, strongly indicating that these enhancer regions are contributing to the phenotypic diversity of prostate cancer. Taken together, these analyses highlight several new patterns of active enhancer regions that associate with specific prostate cancer subtypes. We are integrating eRNA activities with DNA methylation and transcriptome data from the same cell lines to uncover detailed regulatory programs in prostate cancer.
................................................................................................................
RG P46: Viral and retrotransposon sequences have shaped the preferred contexts for APOBEC-mediated mutagenesis
Jeffrey Chen1 , Thomas MacCarthy1
1Stony Brook University
The AID/APOBEC gene family of cytidine deaminases consists of mutagenic enzymes that have evolved roles in innate immunity such as virus restriction and suppression of transposable elements, particularly in mammals. The ancestral APOBEC gene, Activation Induced Deaminase (AID) arose early in vertebrate evolution and plays a key adaptive immunity role (somatic hypermutation of the Immunoglobulin genes) in all jawed vertebrates. Biochemical and in vivo profiling of many APOBECs shows they cause C to T transitions and have evolved a variety of local DNA sequence context preferences. APOBEC3F, for example, has a preference for mutations at TTC sites whereas APOBEC3G has a preference for CCC. We assess the impact of each motif on a set of potential target genes to investigate how individual preferences have been shaped. By specifically examining the impact of replacement mutations we demonstrate that the known APOBEC preferences maximally impact retrotransposons while minimally impacting essential host genes. Furthermore, permutation analysis of several mammalian virus genomes shows these have evolved to avoid the impact of these mutations. Our results also suggest that APOBEC preferences impose restrictions on codon and amino acid usage in their target genes by, for example, heavily disfavoring amino acid pairs that must encode the TTC motif favored by APOBEC3F.
................................................................................................................
RG P47: ATAC-seq is predictive of chromatin state
Chuan-Sheng Foo1 , Sarah Denny1, Jason Buenrostro1, William Greenleaf1, Anshul Kundaje1
1Stanford University
Distinct combinations of chromatin modifications (chromatin states) have been found to be associated with different types of active and repressed functional elements in the human genome such as promoters, enhancers, and transcribed elements. Previously, multivariate hidden Markov models (e.g. ChromHMM and Segway) have been used to learn combinatorial chromatin states and automatically annotate genomes. However, such methods typically require multiple high-quality chromatin mark datasets as input, thus limiting their applicability in practice. Chromatin ChIP-seq experiments are time-consuming and costly to perform, and more importantly, require large amounts of input material to obtain reliable signal. We (Greenleaf lab) recently developed an assay, ATAC-seq, that accurately profiles genome-wide chromatin accessibility, DNA binding protein footprints, and nucleosome positioning from low amounts of input material based on direct in vitro transposition of sequencing adaptors into native chromatin. We previously showed that loci with different chromatin states (learned from histone modification ChIP-seq datasets) showed distinct distributions of ATAC-seq insert sizes in aggregate.
In this work, we further this connection between chromatin architecture and chromatin states by showing that chromatin architecture is in fact predictive of chromatin state at individual loci. More concretely, we show that a machine learning model trained on various features derived solely from ATAC-seq data is able to accurately predict different classes of regulatory elements in active and repressed chromatin states in cell lines and primary cells. The success of our method suggests that different classes of regulatory elements are associated with distinct open chromatin and nucleosome positioning signatures. We explore the feasibility of cross-cell-line chromatin state prediction and determine the minimum sequencing depth required for good predictive performance by subsampling reads. In conclusion, when applied to ATAC-seq data, our method enables high quality genome-wide chromatin state annotations from low quantities of input material using a single assay, potentially enabling the in vivo dissection of chromatin states from (rare) sorted cell populations in primary tissue.
................................................................................................................
RG P48: Identifying genetic and environmental determinants of gene expression
Roger Pique-Regi1 , Christopher Harvey1, Gregory Moyerbrailean1, Omar Davis1, Donovan Watza1, Xiaoquan Wen2, Francesca Luca1
1Wayne State University, 2University of Michigan, Ann Arbor
The effect of genetic variants on a molecular pathway, and ultimately on the individual's phenotype, is likely modulated by "environmental" factors. However, it is generally difficult to determine in which tissues and conditions genetic variants may have a functional impact. We denote the functional genetic variants that show cellular environment-specific effects as GxE expression quantitative trait loci (GxE-eQTLs). Achieving a better understanding of the mechanisms underlying GxE-eQTLs is a critical step in understanding the link between genotype and complex phenotypes.
To identify and characterize GxE-eQTLs we have established a new two-step and cost-effective experimental approach. In the first step, we identify global changes in gene expression using low-coverage sequencing of pools of highly multiplexed samples. In the second step, we select a subset of samples for deep sequencing and allele-specific analysis. For the first step, we generated 960 RNA-seq libraries in pools of 96 spanning 265 cellular environments across 5 cell-types (3 individuals), and 53 different treatments (including hormones, dietary components, environmental contaminants and metal ions). Relevant GO categories were enriched in the observed global gene expression changes (e.g., immune response for Dexamethasone, ion homeostasis for Zinc). We then analyzed allele specific expression (ASE) using a novel method (QuASAR) that allows for joint genotyping and allele specific analysis on RNA-seq data. Across 56 cellular environments we discovered 7738 instances of ASE (FDR<10%), corresponding to 6234 unique ASE genes. Using a Bayesian model across treatments within cell types, we observed that generally >95% ASE signals are shared and their effect sizes are highly concordant (posterior correlation coefficient 0.9). This is highly consistent with previous analysis of condition-specific eQTLs. Nevertheless, out of 112,564 tests we still estimate 2318 loci with a Bayes posterior probability supporting GxE interaction (1273 sites treatment-specific and 1045 sites control-specific, GxE-eQTLs). Genes that are differentially expressed also show a higher enrichment for condition-specific ASE. Our results constitute a first comprehensive catalog of GxE-eQTLs and we anticipate that it will contribute to the discovery and understanding of GxE interactions underlying complex traits.
................................................................................................................
RG P49: MyoD induces active and poised chromatin structures during transdifferentiation
Dinesh Manandhar1 , Lingyun Song1, Ami Kabadi1, Charles Gersbach1, Raluca Gordan1, Greg Crawford1
1Duke University
Overexpression of transcription factor (TF) MyoD has been shown to transdifferentiate cells from non-myogenic lineages into cells with muscle-like expression and functional characteristics. However, expression studies show that the transdifferentiated cells have only some myogenic genes upregulated. Chromatin level reprogramming is also incomplete. In this work, we investigate the reasons behind incomplete MyoD-induced transdifferentiation of fibroblasts, including potential MyoD cofactors, DNA methylation, and posttranslational histone modifications. We analyzed high-throughput chromatin accessibility (DNase-seq) data, in vivo MyoD binding (ChIP-seq) data, and global gene expression (RNA-seq) data on primary skin fibroblast cells transduced with inducible MyoD, and compared against the data obtained from starting fibroblast cells and target myoblasts and myotubes. Our study of local chromatin changes genome-wide suggests that the chromatin state of transdifferentiated fibroblasts is intermediary between fibroblast and muscle chromatin states. Importantly, we observed a continuum of chromatin reprogramming in the MyoD-induced fibroblasts, indicating that complete reprogramming is achieved in only a small fraction of the genome. We also see evidence that during MyoD-induced transdifferentiation, chromatin closes more easily than it opens up. Using random forest and support vector machine classifiers, we show that various genetic and epigenetic features dictate the efficiency of chromatin level reprogramming. For instance, fibroblast DNase hypersensitive sites (DHSs) with higher GC content tend to stay open more than DHSs with low GC content. Our analysis of TF motifs and histone modification data suggests that the presence of certain TFs or histone modification marks at or around a genomic site can dictate the efficiency of chromatin reprogramming. Analysis of gene expression data shows that reprogramming of genes correlates well with reprogrammed chromatin state. Nonetheless, enriched levels of "poised" or "memory" state chromatin are also observed around such genes. This indicates that MyoD is capable of inducing both active and poised chromatin structures that are similar to primary muscle lineages, and that other additional factors - such as Uhrf1, a chromatin remodeler under-expressed in transdifferentiated cells - can potentially help improve the reprogramming efficiency. Interestingly, we also found that although MyoD binding in non-DHSs opens up the chromatin at many genomic loci, a big fraction of MyoD-bound sites remain closed. Most of these closed sites lack MyoD-specific binding sites, which suggests that during transdifferentiation MyoD can also bind non-specifically or mediated by protein cofactors.
................................................................................................................
RG P50: Quantification of DNA cleavage specificity in Hi-C experiments
Dario Meluzzi1, Gaurav Arya1
1University of California, San Diego
Hi-C experiments yield large numbers of DNA sequence read pairs, which are typically analyzed to deduce chromatin interactions across whole genomes. A key step in these experiments is the digestion of cross-linked chromatin with a restriction endonuclease. Although this enzyme is expected to cleave specifically at its recognition sequence, an unknown proportion of cleavages may occur non-specifically, resulting from the enzyme’s star activity or from random DNA breakage. Here we show that Hi-C data sets can be analyzed to quantify such non-specific cleavages. In particular, we describe a computational method to estimate the fractions of cleavages resulting from the putative alternative mechanisms. The method relies on expressing a measured local site distribution near genomic locations of aligned reads as a linear combination of conditional local site distributions. We validated this method using read pairs obtained from computer simulations of Hi-C experiments. We then analyzed a few published Hi-C data sets from murine pre-pro-B and pro-B cells, and found significant variation in cleavage patterns. Knowledge of these patterns may thus enable researchers to optimize Hi-C experimental conditions and fine-tune algorithms for Hi-C data analysis.
................................................................................................................
RG P51: Learning to predict microRNA-mRNA interactions from AGO CLIP-seq and CLASH data
Yuheng Lu1, Steve Lianoglou1, Christina Leslie1
1Memorial Sloan Kettering Cancer Center
MicroRNAs mediate post-transcription gene regulation by guiding the binding of RISC to cognate sites in mRNA transcripts and play critical roles in numerous biological processes. Over the last decade, researchers have mainly focused on canonical rules of microRNA targeting – namely, Watson-Crick pairing between the 5’ seed region of the microRNA and complementary sequences in mRNA targets – but have also reported non-canonical microRNA target sites, which are functional but lack perfect seed pairing. Recently developed high-throughput technologies, like AGO CLIP sequencing and CLASH (crosslinking, ligation, and sequencing of microRNA-RNA hybrids) have made it possible to directly identify a large number of microRNA target sites across the transcriptome. These data underscore the prevalence on non-canonical targets and conversely show that exact microRNA seed matches are not always AGO-bound, indicating that microRNA targeting is determined by factors beyond seed matches. Here we present a novel model for microRNA target prediction based on discriminative learning on transcriptome-wide AGO CLIP and CLASH profiles. As the CLASH protocol captures direct interactions between microRNAs and mRNAs by ligation, it provides a partially labeled microRNA-mRNA pairing dataset, along with the AGO binding sites identified by both AGO CLIP and CLASH. We train support vector machine (SVM) classifiers that model the microRNA-mRNA pairing duplexes and both the local and global context of AGO binding. The duplex and context models together outperform existing target prediction approaches when evaluated on AGO binding and microRNA perturbation expression data sets. Our flexible representation of microRNA-mRNA duplex structures also enables the classifier to predict both canonical and non-canonical pairings between microRNA and target sequences. Moreover, interpretation of the learned models has revealed novel duplex and context features about microRNA targeting. Therefore, this study gives a better characterization of more general microRNA targeting principles and improves target prediction by leveraging rich new high-throughput data with discriminative learning.
................................................................................................................
RG P52: Nencki Genomics Portal – a web-based platform for analysis of transcriptional co-regulation and function, starting from (epi-) genomic and expression data
Michal Dabrowski1, Izabella Krystkowiak1, Michal Petas1, Jaroslaw Lukow2, Norbert Dojer2, Bozena Kaminska1
1Nencki Institute of Experimental Biology, 2University of Warsaw
We present Nencki Genomics Portal (NGP), a website that integrates tools for analysis of gene transcriptional co-regulation and function. It is accessible to a broad biological community via a web browser at http://galaxy.nencki-genomics.org. The NGP tools are separated into four categories — genomic, expression, regulation and function — and are closely integrated, so that the output of one tool can be an input for another (or can be stored).
The genomic tools leverage on Nencki Genomics Database, which extends Ensembl funcgen. The portal provides functionality of genome-wide refinement of regulatory regions, including mapping them to genes, intersecting with other types of regions, intersecting with TFBS motifs, and visualization of these data for specific genes. This makes public data from funcgen (and thus from ENCODE) immediately available to the user alongside his or her own data.
The expression tools provide a typical workflow of analysis of transcriptomics data, from preprocessed gene expression data (genes x conditions) to identification of differentially expressed genes, clustering, and visualization.
The regulation section provides a specialized version of BNFinder that permits analysis of effects of interactions of several regulatory features on gene expression. This tool uses our mammalian model of cis-regulation, updated to take advantage of experimentally identified gene regulatory regions.
The function tools accept a (ranked) list of genes as inputs and provide a unified interface to established tools, including gProfiler, for analysis of functional annotations, such as Gene Ontology, KEGG, and REACTOME.
In addition to the web browser, the local NGD tools are also accessible programmatically, via the standard SOAP/WSDL interface (webservices.nencki-genomics.org), permitting integration into automated analysis pipelines. The middle layer of NGP is based on Taverna Server, which allows us to seamlessly connect to web services and command line tools, and to rapidly deploy new analysis workflows. The NGP architecture permits future tailoring of the portal to users' needs.
................................................................................................................
RG P53: A validated gene regulatory network and GWAS to identify early transcription factors in T-cell associated diseases
Mika Gustafsson1, Danuta Gawel1, Sandra Hellberg1, Aelita Konstantinell1, Daniel Eklund1, Jan Ernerudh1, Antonio Lentini1, Robert Liljenström1, Johan Mellergård1, Hui Wang2, Colm E. Nestor1, Huan Zhang1, Mikael Benson1
1Linköpings UniveristetUniversitet, 2MD Anderson Cancer Center
The identification of early regulators of disease is important for understanding disease mechanisms, as well as finding candidates for early diagnosis and treatment. Such regulators are difficult to identify because patients generally present when they are symptomatic, after early disease processes. Here, we present an analytical strategy to systematically identify early regulators by combining gene regulatory networks (GRNs) with GWAS. We hypothesized that early regulators of T-cell associated diseases could be found by defining upstream transcription factors (TFs) in T-cell differentiation. Time-series expression profiling identified upstream TFs of T-cell differentiation into Th1/Th2 subsets enriched for disease associated SNPs identified by GWAS. We constructed a Th1/Th2 GRN based on integration of expression, DNA methylation profiling, and sequence-based predictions data using LASSO algorithm. The GRN was validated by ChIP-seq and siRNA knockdowns. GATA3, MAF, and MYB were prioritized based on GWAS and the number of GRN predicted targets. The disease relevance was supported by differential expression of the TFs and their targets in profiling data from six T-cell associated diseases. We tested if the three TFs or their splice variants changed early in disease by exon profiling of two relapsing diseases, namely multiple sclerosis and seasonal allergic rhinitis. This showed differential expression of splice variants of the TFs during relapse-free asymptomatic stages. Potential targets of the splice variants were validated based on expression profiling and siRNA knockdowns. Those targets changed during symptomatic stages. Our results show that combining construction of GRNs with GWAS can be used to infer early regulators of disease.
................................................................................................................
RG P54: Weak base-pairing in both seed and 3’ regions reduce RNAi off-targets and enhance si/shRNA designs
Shuo Gu1, Yue Zhang1, Lan Jin1, Yong Huang1, Feijie Zhang1, Michael Bassik1, Martin Kampmann2, Mark Kay1
1Stanford University, 2University of California, San Francisco
The use of RNA interference (RNAi) is becoming routine in scientific discovery and treatment of human disease. However, its applications are hampered by unwanted effects, particularly off-targeting through miRNA-like pathways. Recent studies suggest that the efficacy of such off-targeting might be dependent on binding stability. Here, by testing shRNAs and siRNAs of various GC content in different guide strand segments with reporter assays, we establish that weak base-pairing in both seed and 3’ regions is required to achieve minimal off-targeting while maintaining the intended on-target activity. The reduced off-targeting was confirmed by RNA-Seq analyses from mouse liver RNAs expressing various anti-HCV shRNAs. Finally, our protocol was validated on a large scale by analyzing results of a genome-wide shRNA screen. Compared with previously established work, the new algorithm was more effective in reducing off-targeting without jeopardizing on-target potency. These studies provide new rules that should significantly improve in siRNA/shRNA design.
Top of Page
Updated Nov 5, 2014
SB P01: FAST-SL: An efficient algorithm to identify synthetic lethal reaction/gene sets in metabolic networks
Aditya Pratapa1, Shankar Balachandran1, Karthik Raman1
1Indian Institute of Technology Madras
Synthetic lethal reaction/gene sets are sets of reactions/genes where only the simultaneous removal of all reactions/genes in the set abolishes growth of an organism. In silico, synthetic lethal sets can be identified by simulating the effect of removal of reaction/gene sets from the reconstructed genome-scale metabolic network of an organism. Previous approaches to identifying synthetic lethal reactions in genome-scale metabolic networks have built on the framework of Flux Balance Analysis (FBA), extending it either to exhaustively analyze all possible combinations of reactions, or formulate the problem as a bi-level Mixed Integer Linear Programming (MILP) problem.
FAST-SL circumvents the complexity of both exhaustive enumeration and the bi-level MILP by iteratively reducing the search space and the computational time involved in identification of synthetic lethal reaction sets. FAST-SL, while considering all possible phenotypes and all parts of metabolism, efficiently identifies the targeted phenotypes. Our algorithm shows more than a 4000-fold reduction in search space over exhaustive enumeration of triple lethal sets for Escherichia coli iAF1260 model. Unlike the previous methods used for identification of lethal reaction sets, FAST-SL uses the sparsest solution obtained by solving the flux balance constraints of a metabolic network, which is a linear programming problem, to eliminate reaction combinations that do not lead to a lethal phenotype, thereby reducing the search space for identifying lethal reaction sets.
As our algorithm finds application in the identification of combinatorial drug targets, in this study, we performed synthetic reaction and gene lethality analysis for genome-scale reconstructions of Salmonella enterica typhimurium and Mycobacterium tuberculosis. We validated the reaction lethals obtained using FAST-SL with exhaustive enumeration of reaction deletions up to the order of two for these organisms. The triple lethal reactions obtained for Escherichia coli using FAST-SL have a precise match with the results obtained with exhaustive enumeration, by performing it on a high-performance computer cluster. Our results also completely agree with those of the SL finder algorithm (Suthers, P.F. et al (2009). Mol Syst Biol, 5:301); notably, our algorithm is substantially faster. Further, we also present a mathematical proof for the correctness of our algorithm.
Overall, FAST-SL is a powerful tool to identify the lethal reaction/gene sets, through a massive reduction in the search space over an exhaustive enumeration approach and the SL Finder algorithm. We believe that our algorithm presents an important advance and can enable the rapid enumeration of synthetic lethal reaction/gene sets in genome-scale metabolic networks.
Availability: The MATLAB implementation of our algorithm (compatible with the COBRA toolbox v2.0, a popular toolbox for constraint-based analysis of metabolic networks) is freely available from: https://home.iitm.ac.in/kraman/lab/research/fast-sl.
............................................................................................................................
SB P02: Identification of master regulatory genes as a minimum connected dominating set
Maryam Nazarieh1, Volkhard Helms1
1Saarland University
The identification of gene regulatory networks governing cellular identity is one of the main challenges for understanding the mechanisms underlying cellular differentiation and reprogramming or cancerogenesis. In this work, we reformulate this problem as an optimization problem, namely that of determining a Minimum Connected Dominating Set for directed graphs. Our approach is motivated by the observation that the pluripotency network in embryonic stem cells is maintained by a small set of key transcription factors which share hundreds of target genes. To exactly identify a particular subset of genes among 2n possible subsets of n genes takes exponential time, but approximation algorithms have been developed which find a close to optimal solution in polynomial time with a constant approximation factor. Here, we show on the basis of time-series gene expression data during the cell cycle of S. cerevisiae that this method reliably identifies top regulators that are known to govern the cell cycle in this model organism.
............................................................................................................................
SB P03: Biological signaling pathways and potential mathematical network representations: biological discovery through optimization
Juan Rosas1, Enery Lorenzo1, Lynn Perez1, Michael Ortiz1, Clara Isaza2, Mauricio Cabrera1
1University of Puerto Rico at Mayaguez, 2Ponce School of Medicine and Health Sciences
Establishing the role of different genes in the development of cancer can be a daunting task, starting with the detection of genes that are important in the illness from high-throughput biological experiments. These experiments belong to the ‘omics denomination, as in genomics, proteomics, metabolomics, and the like. Furthermore, it is safe to say that even with a list of potentially important genes it is highly unlikely that these show changes in expression in isolation. A biological signaling path is a more plausible underlying mechanism. This work attempts the analysis of a microarray experiment to build a mathematical network problem. A pre-selection of genes is carried out with a multiple criteria optimization framework previously published by our research group. First results are presented in lung cancer.
............................................................................................................................
SB P04: Discovering disease associated molecular interactions using Discordant correlation
Charlotte Siska1, Katerina Kechris1
1University of Colorado Anschutz Medical Campus
A common approach for identifying molecular features (such as transcripts or proteins) associated with disease is testing for differential expression or abundance in –omics data. However, this approach is limited for studying interactions between molecular features, which would give a deeper knowledge of the relevant molecular systems and pathways. We have developed a method for this purpose that we call the Discordant method. The Discordant method measures the posterior probability that a pair of features has discordant correlation between phenotypic groups using mixture models and the EM algorithm. We compare our method to existing approaches, one that uses Fisher’s transformation in a classical frequentist framework and another that uses an Empircal Bayes joint probability model. We prove with simulations and miRNA-mRNA glioblastoma data from the Cancer Genome Atlas that the Discordant method performs better in predicting related feature pairs. In simulations we demonstrate that while all of the methods have similar specificity, the Discordant method has better sensitivity and is better at identifying pairs that have a correlation coefficient close to 0 in one group and a largely positive or negative correlation coefficient in the other group. Using the glioblastoma data, which has matched samples between miRNA and mRNA, we find that the Discordant method finds relatively more glioblastoma-related miRNAs compared to other methods. We conclude from the results in both simulations and glioblastoma data that the Discordant method is more appropriate for identifying molecular feature interactions unique to disease.
............................................................................................................................
SB P05: The effect of context-specificity on predicting mechanism of action
Yishai Shimoni1, Mukesh Bansal1, Jung Hoon Woo1, Paola Nicoletti1, Charles Karan1, Andrea Califano1
1Columbia University
We recently developed an algorithm (DeMAND) to elucidate the mechanism of action of a compound using gene expression data following compound-perturbation and a regulatory network. An important question is what is the effect of the cell-type-specificity of network on the quality of the predictions? Here we analyze the effect of using various regulatory networks on the same data and benchmark their influence on identifying genes that are known to be involved in the mechanism of action of the tested compounds. We use context-specific networks from various platforms, context independent networks (from the STRING database), and networks that are specific to other cell types. Our results show that context specificity is essential to achieve high performance.
............................................................................................................................
SB P06: Autophagy Regulatory Network – a general resource and its application to analyze bacterial autophagy modulation
Dénes Türei1,2,*, László Földvári-Nagy1,*, Leila Gul1,*, Dávid Fazekas1, Dezső Módos1,2,3, János Kubisch1, Tamás Kadlecsik1, Amanda Demeter1, Katalin Lenti1,3, Péter Csermely2, Tibor Vellai1, Tamás Korcsmáros1,2,4,5,§
* These authors contributed equally to this work.
1 Department of Genetics, Eötvös Loránd University, Pázmány P. s. 1C, H-1117, Budapest, Hungary
2 Department of Medical Chemistry, Semmelweis University, PO Box 260, H-1444, Budapest, Hungary
3 Department of Morphology and Physiology, Faculty of Health Sciences, Semmelweis University, Vas u. 17, H-1088, Budapest, Hungary
4 TGAC, The Genome Analysis Centre, Norwich Research Park, Norwich, UKb
5 Gut Health and Food Safety Programme, Institute of Food Research, Norwich Research Park, Norwich, UK
§ corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Autophagy is a complex cellular process having multiple roles, depending on tissue, physiological or pathological conditions. Major post-translational regulators of autophagy are well known, however, they have not yet been collected comprehensively. The precise and context dependent regulation of autophagy necessitates additional regulators, including transcriptional and post-transcriptional components that are listed in various datasets. Prompted by the lack of systems-level autophagy-related information, we manually collected the literature and integrated external resources to gain a high coverage autophagy database. We developed an online resource, Autophagy Regulatory Network (ARN; http://autophagy-regulation.org), to provide an integrated and systems-level database for autophagy research. ARN contains manually curated, imported and predicted interactions of autophagy components (1,485 proteins with 4,013 interactions) in humans. We listed 413 transcription factors and 386 miRNAs that could regulate autophagy components or their protein regulators. We also connected the above mentioned autophagy components and regulators with signaling pathways from the SignaLink 2 resource. The user-friendly website of ARN allows researchers without computational background to search, browse and download the database. The database can be downloaded in SQL, CSV, BioPAX, SBML, PSI-MI and in a Cytoscape CYS file formats. ARN has the potential to facilitate the experimental validation of novel autophagy components and regulators. In addition, ARN helps the investigation of transcription factors, miRNAs and signaling pathways implicated in the control of the autophagic pathway. The list of such known and predicted regulators could be important in pharmacological attempts against cancer and neurodegenerative diseases. (Turei at al, Autophagy, in press)
Autophagy is also known to be important for intestinal homeostasis and its malfunction is related to inflammatory bowel disease (IBD) and cancer. Conversely, autophagy is often manipulated by intestinal pathogenic bacteria, such as Salmonella. Better understanding the effect of certain bacterial species on the regulation of human intestinal autophagy could help us to propose IBD and colon cancer prognosis markers. Accordingly, we recently examined the potential autophagy regulating functions of 62 protein-protein interactions detected between Salmonella and human cells. We found that at least three of these interactions could have autophagy regulating functions (AvrA-p53; AvrA-Beta catenin; sifA-RAB7A). We also predicted 144 domain-domain based interactions between Salmonella proteins and 106 human proteins involved in autophagy or its regulation (e.g., the autophagy inducing ULK1 and the selective autophagy component ATG16L1). Our domain-motif based prediction found that the Salmonella E3 ubiquitin-protein ligase SlrP could bind to the upstream autophagy regulator PI3K. This interaction is in agreement with the function of SlrP. Thus, these preliminary interaction predictions already show the power of computational biology methods to generate a pool of potential candidates that are responsible for bacterial autophagy modulation.
Keywords: autophagy, regulation, network, protein-protein interactions, transcription factors, signaling pathway, Salmonella, inflammatory bowel disease
............................................................................................................................
SB P07: Cell fate inclination within 2-cell and 4-cell mouse embryos revealed by single-cell RNA sequencing
Fernando Biase1, Xiaoyi Cao1, Sheng Zhong1
1University of California, San Diego
It remains an open question when and how the first cell fate decision is made in mammals. Using deep single-cell RNA-seq of matched sister blastomeres, we report highly reproducible inter-blastomere differences among ten 2-cell and five 4-cell mouse embryos. Inter-blastomere gene expression differences dominated between-embryo differences and noises, and were sufficient to cluster sister blastomeres into distinct groups. Dozens of protein-coding genes exhibited reproducible bimodal expression in sister blastomeres, which cannot be explained by random fluctuations. The protein expression of one, out of four of these bimodal genes tested, Gadd45a, exhibited clear inter-blastomeric contrasts. We traced some of the bimodal mRNA expressions to embryonic genome activation, and others to blastomere-specific RNA depletion. Inter-blastomere differences created co-expression gene networks that were much stronger and larger than those that can be possibly created by random noises. The highly correlated gene pairs at the 4-cell stage overlapped with those showing the same directions of differential expression between inner cell mass (ICM) and trophectoderm (TE). These data substantiate the hypothesis of inter-blastomere differences in 2- and 4-cell mouse embryos, and associate these differences with ICM/TE differences.
............................................................................................................................
SB P08: Inferring disease mechanisms from multiple gene expression datasets
Sahar Ansari1, Michele Donato1, Sorin Draghici1
1Wayne State University
The ultimate goal of any biological experiment is to understand the underlying phenomenon of the investigated condition. Understanding the mechanisms that cause changes in a phenotype requires the identification of the genes that are disrupted in that phenotype, and the relationships between them.
The networks that explain the interactions between genes can be used to 1) predict the disease or the responses of the system to a specific impact (e.g. drugs), and 2) find the subset of genes that interact with each other and have an important involvement in the condition of interest.
Current technologies allow us to measure gene expression with unprecedented accuracy. The interactions between genes can imply an indirect relation between them via their protein product(s) or their transcription factor(s). Another source of information that can help in the understanding of gene-gene interaction is the physical interactions between genes such as protein-protein interactions (PPI) and/or protein-DNA interaction (PDI) networks.
The currently available methods fail to discover the condition-specific relationships between genes with high accuracy. Many existing methods find gene regulatory networks without focusing on one specific phenotype. These networks are not precise, because genes interact with each other differently in different conditions. As an example there are more than 300 interactions in the KEGG pathway database that exist only in phenotype-specific pathways (e.g. Alzheimer’s disease, colorectal cancer, etc.) and do not exist in others.
In this work, we use multiple gene expression data sets to find the effect of the genes on the ones downstream. In the snapshot data the expression of the genes is measured at one time point; therefore, the effect of one gene on the others may not be captured in only one dataset.
We use the union of the differentially expressed (DE) genes from each dataset as a unique list of DE genes. We build a “neighbor” network for each gene with the edges from these genes and all genes immediately downstream of them in the PPI network. In the next step, we calculate the enrichment of each neighbor network based on the number of DE genes they contain. In the last step, we construct a unique network by joining all significant neighbor networks that are connected to each other. This overall network can span different existing pathways or can be a subgraph or one. This network represents the proposed putative mechanism that is consistent with all measured expression changes, all known PPI interactions and are unlikely to be impacted to the level observed just by chance. We applied this approach on three datasets that come from experiments studying type II diabetes. We assessed the result by comparing the constructed network with the pathways that are associated with diabetes in the KEGG database. We rank the pathways based on their enrichment compared to the resulting mechanism. Pathways such as the TGF-beta signaling pathway (enrichment p-values=6.74e-40) and pancreatic secretion pathway (enrichment p-values=4.78e-06), which are associated to type II diabetes, are highly ranked among other pathways. We also performed Gene Ontology (GO) analysis to find the biological processes that are enriched with the resulting mechanism. The results show that the proposed mechanism includes genes known to have important functions in type II diabetes. Also, many of the interactions included in the putative mechanism are present in pathways that are known to be associated with type II diabetes.
............................................................................................................................
SB P09: DSPathNet: a novel computational framework to decipher drug signaling pathway networks for understanding drug action
Jingchun Sun1, Min Zhao2, Peilin Jia3, Lily Wang3, Yonghui Wu1, Carissa Iverson3, Yubo Zhou4, Erica Bowton3, Dan Roden3, Joshua Denny3, Melinda Aldrich3, Hua Xu1, Zhongming Zhao3
1University of Texas Health Science Center at Houston, 2Vanderbilt University School of Medicine, 3Vanderbilt University, 4Chinese Academy of Sciences
A drug performs its function via a cascade to transfer chemical signals from drug binding proteins to signal recipient transcription factors (TFs). The cascade is complicated, involving multiple signaling pathways acting in the network mode. Reconstruction of signaling pathway networks is vital for the identification of drug targets and off-targets, which in turn facilitates our understanding of the mode of drug action and drug development. However, it is challenging to abstract multiple signaling pathways involved in the drug action into one system.
To address this challenge, we developed a novel computational framework, a Drug-specific Signaling Pathway Network method (DSPathNet), for constructing a signaling pathway network (SPNetwork) for an individual drug of interest. The SPNetwork is expected to include genes that harbor genetic variations contributing to the pathology of the drug indication or drug response. We illustrated the utility of DSPathNet using metformin, which is one of the most widely prescribed anti-diabetic drugs in the world and has been recently shown to be useful for cancer treatment and prevention in people at higher risk. Given the available data and the nature of signal transduction cascades, we compiled 65 metformin upstream genes and 66 metformin downstream genes. Then by overlaying them onto the human SPNetwork, we compiled and applied random walk algorithms through longitudinal and lateral movements, generating one metformin-specific SPNetwork with 477 nodes and 1,366 edges. By examining the disease genes and genotyping data of multiple GWAS data in the network, we found that the metformin network was significantly enriched with disease genes for both T2D and cancer, and that the network also included genes that may be associated with metformin-associated cancer survival. Furthermore, from the metformin SPNetwork and common genes to T2D and cancer, we generated a subnetwork to highlight molecule crosstalk between T2D and cancer. The follow-up network analyses and literature mining revealed that seven genes (CDKN1A, ESR1, MAX, MYC, PPARGC1A, SP1, and STK11) and one novel MYC-centered pathway with CDKN1A, SP1, and STK11 may play important roles in metformin’s antidiabetic and anticancer effects.
In this study, we showed that 1) DSPathNet is a novel approach for constructing drug-specific signal transduction networks; 2) Metformin-specific SPNetwork provides insights into the molecular mode of metformin; and 3) the study serves as a model for exploring signaling pathways to facilitate understanding of drug action, disease pathogenesis, and identification of drug targets.
............................................................................................................................
SB P10: Learning nucleotide groups from RNA structure-probing data
Xihao Hu1, Kevin Yip1
1The Chinese University of Hong Kong
Motivation: Genome-wide RNA structure probing has become popular in recent years, especially in the study of RNA structures in vivo. High-throughput methods that detect modifications on unpaired adenines and cytosines by dimethyl sulphate (DMS) treatment are able to distinguish exposed, unpaired nucleotides from others in either in vitro or in vivo systems. Previously we have developed a mixture of Poisson linear model to fit the raw read counts from high-throughput RNA structure probing data based on structure-specific enzymatic cuts. We have shown that the hidden states learned by the model can be combined with other sequence features to predict protein binding sites on RNAs.
Results: We have applied our developed methods to new DMS data. We compared the modeling results from data obtained by three different conditions, namely in vivo, in vitro, and denatured. We found that our method provided the highest improvements of modeling accuracy as compared to other simpler models when applied to in vivo data. The results suggest that DMS data from in vivo systems are closer to a mixture of states, which we hypothesize to be a state for exposed unpaired nucleotides and a state for all other nucleotides. By transforming raw read counts into probabilities of the two states, our method makes the sequencing data easier to interpret and provides useful features for downstream analyses. We have made our model and software publicly available, and hope it can help extract important features of various types of RNA structure probing data.
............................................................................................................................
SB P11: Expression amplitude based drug repositioning
Mario Failli1, Vincenzo Belcastro1
1Telethon Institute of Genetics and Medicine (TIGEM)
Background: Traditionally, most drugs have been discovered using phenotypic or target–based screens, but their indications are often expanded on the basis of clinical observations, providing additional benefit to patients. For that reason and in response to the high cost and risk in traditional de novo drug discovery, discovering potential uses for existing drugs, also known as drug repositioning, has attracted increasing interest from both the pharmaceutical industry and the research community (Hurle et al., 2013).
Recent research has shown that computational approaches have the potential to offer systematic insights into the complex relationships among drugs, targets, and diseases for successful repositioning. In particular, targeted mechanism-based drug-repositioning methods integrate treatment omics data to delineate the unknown mechanisms of action of drugs. Such research led to the development of computational approaches to predict drug mode of action (MoA) and drug repositioning from the analysis of the Connectivity MAP (CMAP) (Lamb J. et al., 2006), a compendium of gene expression profiles (GEPs) following drug treatment of 5 human cell lines with 1309 bioactive small molecules.
Iorio et al. (2009) first proposed to construct a ‘Prototype List’ of the drug by merging its experiments from cell lines, batches, concentrations, and microarray platforms. A following study by Iskar et al. (2010) overcame the batch effect (Lander E.S., 1999) by implementing a novel protocol with filtering and normalization steps, applicable to gene expression upon heterogeneous drug treatments. However, both methods suffer from two main limitations: lack of confidence levels over the ranked lists of genes, and use of fixed-length prototype ranked lists to compute drug-drug associations.
Objective: Development of a procedure to associate to each drug the full list of differentially expressed genes, following treatment across multiple cell lines or different dosages, while keeping information on the ratio between treatment and control (fold change) and its statistical significance (p-value). Both fold changes and p-values are then combined to compute drug-drug associations over the full list of genes.
Material/Methods: The approach requires two normalization steps: first, a Robust Multi-array Averaging (RMA) algorithm (Gautier et al., 2004) normalizes between treated and untreated samples of a single experiment. Second, the ‘quantile’ normalization (Yang and Thorne, 2003) forces the RMA normalized arrays, belonging to the same drug, to have identical empirical distribution in order to avoid batch effects. A linear model of treated conditions versus controls is then fitted, and the significance level of each differentially expressed gene computed. Probesets are then ranked based on a combination of both fold change and p-value (Martin et al., 2012), and pairwise distances between drugs computed via Spearman rank correlation. A similar approach was applied to derive pairwise drug correlations from Iorio and Iskar’s ranked lists for comparison purposes.
Results and Discussion: We collected for 469 out of 511 drugs the Anatomical Therapeutic Chemical (ATC) code (Pahor et al., 1994) and determined their distances adopting string similarity criteria. We next compared the three methods by computing the Kolmogorov-Smirnov (KS) statistic to check whether drugs sharing high Spearman correlations tend to be close in terms of ATC distance. Although all three methods performed better than random, the data collected from our method indicated the highest significance (with picks of >10 orders on KS significance levels) on a wide range of correlation intervals. Hence the method is a valid resource for drug repositioning; in addition, the presence of significance levels over lists of genes ease the integration with other bioinformatics resources (i.e., coexpression gene networks) to improve predictions.
............................................................................................................................
SB P12: A systems biology approach highlights the role of GSK3-beta in the regulation of PDX1 by IL1-beta in pancreatic beta cell
Jisha Vijayan1, Yogeshwari Sivakumaran1, Rajagopal Rangarajan1, Mahesh Verma1, Anup Oommen1, Krishnamurthy Sheshadri1
1Connexios Life Sciences
A network model describing the response of proliferation (PDX1) to inflammation (IL1-Beta) in beta cell is described. The model is automatically extracted using an automated path-tracing algorithm from a large beta cell network based on integration of extensive literature. A mathematical model based on mass action kinetics is formulated for this network model. A steady-state simulation of the mathematical model shows that PDX1 decreases as IL1-beta is increased, saturating at high IL1-beta levels. Measurements on mouse pancreatic beta cell line (NIT-1) confirmed this behavior. Further simulations showed that under conditions of GSK3-beta inhibition, the response of PDX1 to IL1-beta reverses to increasing behavior, again saturating at high IL1-beta levels; further, the PDX1 levels were lower in this case. The saturation levels in both of these cases are comparable. Again this behavior was confirmed by measurements. This study highlights the role of GSK3-beta in the switching of PDX1 response to IL1-beta.
............................................................................................................................
SB P13: Elucidating complex phenotypes based on high-throughput expression and biological annotation data
Nitesh Singh1, Mathias Ernst1, Volkmar Liebscher2, Georg Fuellen1, Leila Taher1
1University of Rostock, 2Ernst Moritz Arndt University of Greifswald
The interpretation of large gene expression datasets describing complex phenotypes is obscured by the fact that such phenotypes are likely to be controlled by the concerted action of multiple genes. Combining gene expression analysis with annotation regarding the functions, processes, and pathways in which the genes are involved has the potential to elucidate the biological interactions underlying a given phenotype. Here, we present an approach that integrates gene expression and biological annotation data to identify biological units and their interactions that influence a phenotype of interest. First, we divide genes with similar biological annotation into clusters. Second, we group the genes within each cluster into sub-clusters, based on their expression profiles. Finally, we construct a co-expression network of sub-clusters to analyze the interactions between the biological units represented by these sub-clusters. We applied our approach to two microarray expression datasets describing the differentiation of mouse embryonic stem cells into embryoid bodies, and mouse liver development and regeneration, respectively. For the first dataset, our findings confirm that developmental processes and apoptosis have a key role in cell differentiation. Furthermore, we suggest that processes related to pluripotency and lineage commitment, which are known to be critical for development, interact mainly indirectly, through genes implicated in more general biological processes. For the second dataset, we concluded that the transcriptional mechanisms beneath liver regeneration are fundamentally different from those regulating embryonic liver development. Moreover, we provide evidence that supports the relevance of cell organization in the developing liver for proper liver function. Understanding how genes involved in specific biological functions, processes, and/or pathways interact depending on particular experimental conditions is crucial to decipher the molecular basis of health, disease, and drug response. This study provides a new approach to examining gene expression data that can be easily extended to other high-throughput expression data.
............................................................................................................................
SB P14: Preserving biological heterogeneity with a permuted surrogate variable analysis for genomics batch correction
Hilary Parker1, Jeffrey Leek1, Alexander Favorov1, Xiaoxin Xia2, Sameer Chavan1, Christine Chung1, Elana Fertig1
1Johns Hopkins University, 2Rutgers University
Sample source, procurement process, and other technical variations introduce batch effects into genomics data. Algorithms to remove these artifacts enhance differences between known biological covariates, but also carry potential concern of removing intragroup biological heterogeneity and thus any personalized genomic signatures. As a result, accurate identification of novel subtypes from batch-corrected genomics data is challenging using standard algorithms designed to remove batch effects for class comparison analyses. Nor can batch effects be corrected reliably in future applications of genomics-based clinical tests, in which the biological groups are by definition unknown a priori. Therefore, we assess the extent to which various batch correction algorithms remove true biological heterogeneity. We also introduce an algorithm, permuted-SVA (pSVA), using a new statistical model that is blind to biological covariates to correct for technical artifacts while retaining biological heterogeneity in genomic data. This algorithm facilitated accurate subtype identification in head and neck cancer from gene expression data in both formalin-fixed and frozen samples. When applied to predict human papillomavirus (HPV) status, pSVA improved cross-study validation even if the sample batches were highly confounded with HPV status in the training set.
............................................................................................................................
SB P15: Experimental design for regulatory network discrimination
Jukka Intosalmi1, Henrik Mannerstrom1, Harri Lähdesmäki1,2
1Aalto University, 2Turku Centre for Biotechnology, University of Turku and Åbo Akademi
Biochemical systems such as regulatory networks can in many cases be modeled using ordinary differential equations (ODEs). The construction of ODE models is typically based on some initial information on interactions between different components and known patterns of stationary or temporal behavior. Typically, neither the interactions nor the parameter values defining the interaction strengths are fully known and have to be inferred from available or forthcoming data. During the past decades, numerous statistical methods for parameter inference and model selection have been developed to carry out this challenging task.
While both parameter inference and model selection for regulatory networks are studied extensively, the development of experimental design methods that can be used to predict the most useful and efficient experiment for model selection has gained much less attention.
In this work, we present a novel procedure to design optimal experiments for model selection of regulatory network models. Our approach relies on a utility-based Bayesian framework that enables the efficient use of prior information not only on the parameter values but also over the dynamics of model responses. We exemplify the usefulness of our approach by constructing an optimal experimental design (of measurement time-point selection) for a network inference problem, in the presence of very scarce initial data. In addition, we discuss an efficient numerical implementation of our method and outline promising future applications.
............................................................................................................................
SB P16: Bioconductor's EnrichmentBrowser: Seamless navigation through combined results of set-based and network-based enrichment analysis
Ludwig Geistlinger1, Gergely Csaba1, Ralf Zimmer1
1Ludwig-Maximilians-Universität Munich
Background: Enrichment analysis of gene expression data is essential to find functional groups of genes whose interplay can explain experimental observations. Numerous methods have been published that either ignore (set-based) or incorporate (network-based) known interactions between genes. However, the often subtle benefits and disadvantages of the individual methods confusing for most biological end users and there is currently no convenient way to combine methods for an enhanced result interpretation.
Results: We present the EnrichmentBrowser package as an easily applicable software that enables (1) the application of the most frequently used set-based and network-based enrichment methods, (2) their straightforward combination, and (3) a detailed and interactive visualization and exploration of the results. The package is available from the Bioconductor repository and implements additional support for standardized expression data preprocessing, differential expression analysis, and definition of suitable input gene sets and networks.
Conclusion: The EnrichmentBrowser package implements essential functionality for the enrichment analysis of gene expression data. It combines the advantages of set-based and network-based enrichment analysis in order to derive high-confidence gene sets and biological pathways that are differentially regulated in the expression data under investigation. Besides, the package facilitates the visualization and exploration of such sets and pathways.
............................................................................................................................
SB P17: CoRegNet: reconstruction and interrogation of co-regulatory network
Rémy Nicolle1, François Radvanyi2, Mohamed Elati1
1Institute of Systems & Synthetic Biology, Université d'Evry, 2Institut Curie
Recent advances in large-scale transcriptomics have enabled the profiling of hundreds to thousands of tumor samples by large consortia such as the The Cancer Genome Atlas (TCGA: http://cancergenome.nih.gov) or the International Cancer Genome Consortium (www.icgc.org). While the amount of data holds great promise for our understanding of tumor progression, they necessitate new and efficient methodologies to be analyzed and to extract valuable knowledge from them.
We present COREGNET, an R/Bioconductor package (under revision) that implements a set of tools to infer and analyze co-regulatory networks from gene expression data. The functions implemented in the package are based on previously validated and published studies. The first step of the proposed workflow is based on H-LICORN (1,2), a hybrid method network inference method using both a discretized and continuous version of the data to infer the set of cooperative regulators of genes. In order to improve the predicted network, the package implements methods derived from the modENCODE project (3) to refine the predictions by integrating additional regulatory evidence from data such as transcription factor binding sites, ChIP-seq, or ChIP-on-chip. The second step of the workflow aims at identifying, based on transcriptomic data, the combination of active transcription factors in a given sample. Based on our studies of feature extraction in gene expression (3), the proposed method uses the structure of the network to estimate the activity of a given transcription factor in a given sample through the expression of its target genes. This transformation of the data results in a new dataset representing the whole transcriptome of every sample by the activity of transcription factors. Additional implemented functions are derived from MARINa (5) (MAster Regulator Inference algorithm) to identify the most specific regulator of a given set of target genes of interest. Finally, an embedded shiny application eases the analysis of the co-regulatory network, containing links between cooperative regulators, through an interactive display of the network using a Cytoscape applet (javascript, non flash-based) integrating gene expression, transcription factor activity, and other features such as mutations or copy number alteration.
References
1. I. Chebil, R. Nicolle, G. Santini, C. Rouveirol and M. Elati. Hybrid method inference for the construction of cooperative regulatory network in human, IEEE transactions on nanobioscience, 13: 97 - 103, 2014.
2. Elati M, Neuvial P, Bolotin-Fukuhara M, Barillot E, Radvanyi F. and Rouveirol C. LICORN: learning co-operative regulation networks from expression data. Bioinformatics, 23:2407-2414, 2007.
3. Marbach D, Roy S, Ay F, Meyer PE, Candeias R, Kahveci T, Bristow CA & Kellis M (2012) Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Research 22: 1334–1349.
4. Nicolle R, Elati M & Radvanyi F (2012) Network Transformation of Gene Expression for Feature Extraction. Machine Learning and Applications (ICMLA’11), IEEE 1:108-113.
5. Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, Basso K, Beltrao P, Krogan N, Gautier J, Dalla-Favera R & Califano A (2010) A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Molecular Systems Biology 6: 1–10.
............................................................................................................................
SB P18: BioTapestry: new Version 7 features improve scalability and flexibility
Suzanne Paquette1, Kalle Leinonen1, William Longabaugh1
1Institute for Systems Biology
BioTapestry is a well-established tool for building, visualizing, and sharing models of gene regulatory networks (GRNs), with particular emphasis on the GRNs that drive development. It uses a hierarchy of models to present multiple views of the network at different levels of spatial and temporal resolution, and uses a visual representation that is tailored to the presentation of GRNs. Given their complexity, it is important to provide online interactive tools that can be used to explore a GRN model, and the existing Java-based BioTapestry Viewer has been used, for example, to provide an interactive online version of the sea urchin endomesoderm network since 2003.
However, current web-browser technologies such as HTML5 Canvas make it possible to provide an interactive graphical network model directly in a web browser without needing Java; we have now created a version of the BioTapestry Viewer using these technologies. At the same time, BioTapestry's new dual-mode software architecture continues to support the traditional Java-based BioTapestry Editor desktop application. This new feature is part of the new BioTapestry Version 7, which is scheduled for release in the autumn of 2014.
Version 7 also continues to build upon the helpful automatic network layout tools that were introduced in the current Version 6. In particular, we have improved the layout performance, and have also applied lessons learned from developing the new companion BioFabric network visualization tool to enhance BioTapestry’s presentation of large directed networks. These performance improvements are particularly noteworthy, since BioTapestry's "circuit trace" presentation style for directed links is highly scalable, and thus the user can automatically create rational, understandable, and highly organized presentations of large directed networks containing thousands of nodes.
............................................................................................................................
SB P19: Network Infusion to infer information sources in networks
Soheil Feizi1, Ken Duffy2, Muriel Medard1, Manolis Kellis1
1Massachusetts Institute of Technology, 2Hamilton Institute
Several models exist for diffusion of signals across biological, social, or engineered networks. However, the inverse problem of identifying the source of such propagated information seems on the surface intractable, even in the presence of multiple network snapshots, and especially for the single-snapshot case, given the many overlapping paths in real-world networks. Mathematically, this problem can be undertaken using a diffusion kernel that represents diffusion processes in a given network, but computing this kernel is generally intractable.
Here, we introduce a modified diffusion kernel that relaxes the path-coupling constraints by only considering k independent shortest paths among pairs of nodes, assuming an exponential time distribution for node-to-node spreading. We use the resulting Erlang network diffusion kernel to solve the inverse diffusion problem using both likelihood maximization and error minimization. We apply this framework for both single-source and multi-source diffusion, for both single-snapshot and multi-snapshot observations, and using both uninformative and informative prior probabilities for candidate source nodes.
We apply Network Infusion (NI) to identify disease-causing genes of several human diseases including T1D, Parkinson’s, MS, SLE, CVD, CAD, psoriasis, and schizophrenia, and show that NI infers candidate disease-causing genes that are biologically relevant and often not distinguishable using the raw p-values. In a second application, we identify the news sources for 3553 stories in the Digg social news network, and validate our results based on annotated information that was not provided to our algorithm. We also apply NI to several synthetic networks and compare its performance to centrality-based and distance-based methods for Erdos-Renyi graphs, power-law networks, symmetric grids, and asymmetric grids.
We also provide proofs that under a standard susceptible-infected (SI) diffusion model, (1) the maximum-likelihood Network Infusion method is mean-field optimal for tree structures or sufficiently sparse Erdos-Renyi graphs, (2) the minimum-error algorithm is mean-field optimal for regular tree structures, and (3) for sufficiently-distant sources, our multi-source solution is mean-field optimal in the regular tree structure.
............................................................................................................................
SB P20: Discovering patterns in leukemia — from local protein networks to global pathway utilization
Chenyue Hu1, Steven Kornblau2, Amina Qutub1
1Rice University, 2MD Anderson Cancer Center
Acute myeloid leukemia (AML) is a notoriously heterogeneous disease. Molecular variations in AML patients, including both genetic mutations and post-translational events, make targeted therapies extremely difficult. Delineating clinically impactful patient subpopulations and characterizing their disease mechanisms will not only improve diagnosis, it will also open a door to discovering new drug targets. However, most AML studies have focused on a handful of genetic mutations, which fail to fully account for the diversity of AML phenotypes and offer limited therapeutic opportunities. In this study, we developed a computational methodology to analyze protein and phosophoprotein states from AML patient samples. We applied our approach to discover new AML patient subpopulations, characterize local protein networks, build global pathway utilization maps, and tie the molecular patterns back to the clinical information and outcome of patients.
Profiles of 231 protein expression levels in 560 AML patient bone marrow samples as well as 21 normal bone marrow samples were obtained using Reverse Phase Protein Array (RPPA). Based on prior knowledge of protein functionality, we first grouped all the proteins into 24 pathways (e.g., hypoxia, adhesion, apoptosis). This categorization allows us to explore patient subpopulations and network patterns in a better-defined biological context. For each pathway, we then clustered patients into groups with distinct protein expression patterns using a combination of prototype clustering (estimating optimal cluster number) and K-means. We observed patterns that are consistent with normal biological mechanisms, as well as patterns that can not be explained by our current understanding of cell biology. To distinguish disease-specific patterns, we mapped normal samples onto patient samples in reduced dimensions of protein expression levels. Normal samples overlapped significantly with patient clusters in some pathways (e.g., differentiation, cell cycle), while they appeared distinct from almost all patient clusters in other pathways (e.g., heatshock, ribosome). We also identified patient clusters that are prognostic (e.g., adhesion) and clusters that are strongly associated with certain clinical correlates (e.g., cell differentiation). To investigate protein interactions, we built local protein networks combining both interactions from public databases (e.g., KEGG, STRING) and interactions inferred from the data set using glasso. These networks demonstrated a large contrast between what is known in literature and what is suggested from the new clinical data. In addition, we organized patterns in each pathway into a pathway activation roadmap, which illustrates multiple potential routes cells can hijack to activate or deactivate a pathway. At the global level, we translated all pathway patterns into a barcode system and were able to identify pathway groups that are co-utilized by different subpopulations.
In sum, results of this proteomic analysis uncovered potential pathways to target therapeutically and to use as biomarkers for groups of leukemia patients. At the same time, it provides a computational strategy that can be applied broadly to clinical omics data. Furthermore, the global analysis reconstructed a molecular utilization map for AML — one that could be a key to unlocking mechanisms of cancer.
............................................................................................................................
SB P21: A visual exploration of biological networks based on edge-centric view and edge centralities
Divya Mistry1, Julie Dickerson1
1Iowa State University
In a biological network, nodes typically represent the biological entities — genes, proteins, metabolites, or other biomolecules — resulting from experimental data, while edges represent a statistical or biophysical relationship between the nodes. A key problem is prioritizing the potential edges between the nodes for focused investigation of biological networks. To enable visual data mining, this work develops a parallel coordinate plot (PCP) based linear network visualization tool, called VisPNet. In a VisPNet diagram, each axis represents edge properties of interest, such as edge weights, edge centrality measures, or a user-defined metric. VisPNet provides the following advantages for visually mining a biological network.
• By organizing a viewport based on the edge distribution, topological features originating from edge properties become prominent. Because biological networks tend to have more edges than nodes, this organization results in an uncluttered and less noisy view of the network.
• VisPNet provides an option to group or highlight edges based on pre-calculated edge metadata such as weights, functional annotations, cellular localization annotations, and properties of adjacent nodes.
• VisPNet allows interactive brushing along the axes to explore subgraph topologies reliant on the metadata chosen for each of the axes.
• VisPNet can be used for dynamic graph exploration and analysis. Individual networks of various time points or experimental conditions may be laid out as the PCP axes. The ordering of nodes (if relevant), edges, or axes can help highlight consistently similar or divergent biological subnetworks. VisPNet also provides an option to choose up to three axes to generate a static Hive Plot [1], which may help highlight topological patterns.
The current implementation of VisPNet is developed using the D3 JavaScript library [2] atop all the modern browsers with HTML5 capabilities. VisPNet will be available at http://git.io/vispnet in the near future.
References
1. Krzywinski M, Birol I, Jones SJ, Marra MA (2011) Hive plots--rational approach to visualizing networks. Briefings in Bioinformatics: bbr069–. doi:10.1093/bib/bbr069.
2. Bostock M, Ogievetsky V, Heer J (2011) D³: Data-Driven Documents. IEEE Transactions on Visualization and Computer Graphics 17: 2301–2309. doi:10.1109/TVCG.2011.185.
............................................................................................................................
SB P22: A novel model to combine clinical and pathway-based transcriptomic information for the prognosis prediction of breast cancer
Lana Garmire1
1University of Hawaii Cancer Center
With the increasing awareness of heterogeneity in breast cancers, better prediction of breast cancer prognosis is much needed for more personalized treatment and disease management. We here then have developed a novel computational model for breast cancer prognosis by combining the pathway deregulation score (PDS) based pathifier algorithm, Cox regression, and L1-LASSO penalization method. The resulting prognosis genomic model successfully differentiated relapse in the training set (log rank p-value = 6.25e-12) and three testing data sets (log rank p-value < 0.0005), and consistently performed better than gene-based models. Moreover, combining genomic information with clinical information improved the p-values of prognosis prediction by at least three orders of magnitude in comparison to using either genomic or clinical information alone.
............................................................................................................................
SB P24: Verification of biological network models using a collaborative platform
Stephanie Boue1, Anselmo Di Fabio2, Brett Fields3, William Hayes3, Julia Hoeng1, Jennifer Park3, Manuel Peitsch1, Walter Schlage1, Marja Talikka1
1Philip Morris International, 2Applied Dynamic Solutions, 3Selventa
The sbv IMPROVER [Industrial Methodology for PROcess VErification in Research] network verification challenge (NVC) aims to verify and enhance comprehensive biological network models using a web application that facilitates collaboration among the scientific community. These network models are constructed using a structured syntax (Biological Expression Language, BEL) and are supported by scientific evidences at the edge level. We describe here an approach for biological network construction that synergizes manual curation and data-derived components with the process of crowdsourcing. By implementing a reputation-based web application available to the entire scientific community to add biology and vote on supporting scientific evidence, the NVC has created a collaborative crowdsourcing platform to complement peer review of publications describing the networks and to ensure complete and accurate biological networks that can be used as a standard in the field.
A collection of 49 biological network models capture a wide range of biological processes (from cell fate to cell stress, inflammation, and tissue repair) and represent a novel resource to investigate key downstream effectors linking experimental perturbations to specific biological pathways, enabling a predictive analytics pipeline to interpret experimental findings on a mechanistic level. The network models are freely available for the scientific community to download, utilize, and continue to refine for use in toxicological and drug discovery applications.
............................................................................................................................
SB P25: Deconfounding time-series by pseudotemporal estimation
John Reid1, Lorenz Wernisch1
1MRC Biostatistics Unit, Cambridge
Many biological systems of interest evolve over time. Classical examples are the cell cycle and expression patterns in the embryo. Studies of these systems are often cross-sectional in nature. That is, samples taken at distinct time points do not come from the same cells. In many systems, the cells are not synchronized. They progress at different rates and this can confound analyses of such cross-sectional data.
We present a method designed to control for this effect. Our method relies on a probabilistic model which estimates a pseudotime for each sample. On one hand, the pseudotime is related to the observed sample time, on the other hand the measured biological variables are softly constrained to vary smoothly, encouraging the model to explore pseudotimes consistent with the data.
Our model can be viewed as a Gaussian process latent variable model where we place a structural prior on the latent pseudotemporal space. In this sense, it can be seen as a generalization of the work of Buettner and Theis. Our model is also related to the work of Trapnell et al., who use a dimensionality reduction method combined with a maximum-likelihood type estimation procedure to estimate pseudotimes.
We give results for our model showing how it successfully reconstructs pseudotimes in synthetic data and an analysis of data from the cell cycle. The method is applicable to data from populations of cells but is particularly relevant to studies in single cells.
............................................................................................................................
SB P26: Differential regulatory mechanisms: An application to large-scale high throughput molecular profiling of schizophrenia brains
Thanneer Malai Perumal1
1Sage Bionetworks
Dysfunctional networks in disease: High throughput (HTP) molecular profiling data has become a ubiquitous tool for studying a wide range of human diseases. The majority of the existing approaches look for genes that are differentially (co-) expressed. It has now become increasingly important to know how the regulatory networks change along with the change in (co-)expression. Identifying changes in regulatory networks are expected to provide insights into dysregulated mechanisms and to identify key molecular players that may serve as candidate biomarkers or drug targets. To this end, based on HTP RNA-seq and genotype profiling, this work presents a novel ensemble methodology, named differential analysis of regulatory networks (DARN), to elucidate the underlying dysregulated mechanisms in cellular networks resulting in the observed phenotype of disease states.
Detecting differential regulatory networks: Grounded on the principles of perturbation and information theory, DARN uses an ensemble learning approach to infer regulatory interactions from a pool of interactions that are consistent with the expression data and identify mechanisms that differ between cases and controls. Starting from HTP RNA-seq and genotype data, DARN generates regulatory network models using both the data and text mining based approaches. Later, DARN curates the resulting interaction pool using a genetic optimization based ensemble algorithm that enriches the network for the most relevant positive feedback loops for the case-control difference. The major assumption behind this methodology is that the disease and control states are considered as stable expression patterns. Therefore, enriching positive feedback loops, which are a necessary condition for multi-stability, is expected to shed light on disease-induced dysfunction.
Application to HTP datasets of schizophrenia brains: Efficacy of this methodology is showcased through an application to a large-scale HTP data (RNA-seq & genotyping) from the dorsolateral prefrontal cortex (BA9/46) of human post-mortem brain in 265 schizophrenia cases and 289 controls. This dataset was generated in the framework of the CommonMind Consortium (commonmind.org), which aims to generate and analyze large-scale data from human subjects with neuropsychiatric disorders. After normalizing via voom and correcting for the effects of known clinical (gender, age of death, medications) and technical (brain bank, post-mortem interval, RNA quality, sequencing batch) covariates, the initial interaction pool were generated using data-driven methods including CLR, VBSR, TIGRESS, and GENIE3, as well as the literature based text mining toolkit of MetacoreTM. Using this pool of interactions DARN learned a population of regulatory networks representing the changes between control and schizophrenia postmortem brains. Analysis of the resulting population of networks is expected to provide a system level understanding of the genotype-phenotype relationships and key components in schizophrenia regulation, and will generate experimentally testable hypotheses for schizophrenia susceptibility and progression.
............................................................................................................................
SB P27: Variability in B-vitamin dependencies in the human microbiome genomes
Matvei Khoroshkin1 , Andrei Osterman2, Dmitry Rodionov2
1Institute for Information Transmission Problems, Russian Academy of Sciences, 2Sanford-Burnham Medical Research Institute
B vitamins are biochemical cofactors essential for any living systems. Human microbiota is the complex and dynamic community of commensal, symbiotic and pathogenic microorganisms that are present on and within the human body and has an enormous impact on humans. We investigate the ability of bacteria from human microbiome to produce and salvage B vitamins. We have selected the reference set of 1143 bacterial genomes from 7 phyla out of those sequenced in course of Human Microbiome Project (HMP). By using the metabolic subsystems approach (as implemented in the SEED database) and analyzing genomic context and regulons, we have reconstructed biochemical pathways for synthesis of eight B vitamins (thiamin, riboflavin, niacin, biotin, pyridoxine, cobalamin, pantothenate, folate) and predicted putative vitamin transporters in the reference HMP genomes. Using the reconstructed metabolic pathways, we have classified the HMP organisms with respect to their B-vitamin proto-, auxotrophy and their vitamin transport capabilities. The preferable patterns of vitamin dependency were attributed to a number of taxonomic units. For instance, the Bacteroides are mostly prototrophs that are capable synthesizing all B vitamins, excluding cobalamin. On the contrary, the Lactobacillales are auxotrophes for all vitamins, excluding folate. The reference HMP genomes show a relatively high level of conservation of vitamin synthesis phenotypes at the genus level, hence only 25% of the studied genera demonstrate variability of phenotypes for individual vitamins. Also we have identified patterns of vitamin dependency for a number of body sites. Gastrointestinal tract generally shows the prevalence of vitamin prototrophic bacteria, whereas oral cavity, urogenital tract and blood are largely populated by vitamin auxotrophs. This work is important for understanding the role of B-vitamins in maintaining homeostasis of human microbiome community structures and for future developing of specific vitamin diets.
............................................................................................................................
SB P28: Master regulators of luminal and basal subtypes of breast cancer
Archana Iyer1 , Celine Lefebvre2, Yishai Shimoni1, Mukesh Bansal1, Mariano Alvarez1, Jose Silva3, Andrea Califano 1
1Columbia University, 2Gustave Roussy, 3Mount Sinai Medical Center
Breast cancer is a heterogeneous group of diseases that can be stratified into several subgroups based on their molecular signature. Understanding the regulators of these molecular subtypes will allow us to make them more amenable for targeted therapies or personalized medicine. Here we present our discovery of master regulators that are important in the transcriptional regulation of the two major subtypes: basal and luminal. We reverse engineered a breast-cancer specific transcriptional network using large-scale gene expression datasets in breast cancer (TCGA, Metabric, UNC-300) to create a breast cancer interactome. Using MARINa (Master Regulator Inference Algorithm) we have identified specific transcription factors that regulate basal and luminal subtypes of breast cancer. We further validated these master regulators experimentally by performing a pooled shRNA screen on six independent cell lines (2 luminal, 3 basal, and one normal mammary epithelial cell line). The pooled shRNA screen was sampled at days 0, 10, 18, and 25, and genomic DNA was barcoded and sequenced using the Illumina MiSeq technology. Both computational predictions and our experimental results from the deconvolution of the shRNA screen validate the luminal transcription factors (FOXA1, ESRI, and GATA3). In addition, we discover novel luminal-specific transcription factors like TFAP2C. For the basal subgroup, while we discover novel regulators we also find that this group is more heterogeneous compared to the luminal. Importantly we can effectively target this subgroup with a combination of master regulators.
............................................................................................................................
SB P29: Network modeling reveals key features of epithelial-to-mesenchymal transition dynamics in liver cancer invasion
Steven Steinway1 , Jorge Gomez Tejeda Zañudo2, Thomas Loughran1, Reka Albert2
1University of Virginia, 2Penn State University
............................................................................................................................
SB P30: Trafficking and signaling interplay modeling after serotonin receptor activation
Aurélien Rizk1 , Mauno Schelb1, Milica Bugarski1, Maysam Mansouri1, Gebhard Schertler1, Philipp Berger1
1Paul Scherrer Institute
Despite the physiological and pharmacological importance of G protein-coupled receptors (GPCRs), receptor activation and its translation into cytoplasmic trafficking and cellular response remain elusive. In this project, we study the interplay between signaling and trafficking of serotonin receptors 5-HT2c after stimulation. We use RAB GTPases as markers of intracellular compartments to monitor the dynamic distribution of receptors after stimulation and ERK phosphorylation to monitor signaling output. In order to obtain statistically significant trafficking data and high temporal resolution we developed the "Squassh" image analysis software for automatic vesicles segmentation, counting, and colocalization computation [Rizk et al., Nature Protocols 2014]. Based on the receptor localization data, signaling data and previous work on the modeling of GPCR activated signaling pathways [Heitzler et al., MSB 2012] we developed an ordinary differential equation model combining signaling with receptor internalization and transport to early, recycling, and late endosomes. This is to our knowledge the first attempt to develop a dynamic trafficking model for a GPCR. We evaluate trafficking influence on signaling by conducting global sensitivity analysis and use the model to test hypotheses on receptor constitutive internalization, trafficking regulation, and signaling from endosomes.
............................................................................................................................
SB P31: Understanding multicellular function and disease with human tissue-specific networks
Arjun Krishnan1 , Casey Greene2, Aaron Wong1, Emanuela Ricciotti3, Rene Zelaya2, Daniel Himmelstein4, Daniel Chasman 5, Garret Fitzgerald3, Kara Dolinski1, Tilo Grosser3, Olga Troyanskaya1
Princeton University, 2Dartmouth College, 3University of Pennsylvania, 4University of California, San Francisco, 5Harvard Medical School
Tissue and cell-type identity lie at the core of human physiology and disease. Therefore, understanding the genetic underpinnings of complex tissues and individual cell lineages is crucial for developing improved diagnostics and therapeutics. Yet we still lack tools to systematically explore the landscape of genes and interactions that shape specialized cellular functions across hundreds of tissue types and cell lineages in the body. Here we present genome-wide functional interaction networks specific for each of 144 human tissues and cell types developed using an integrative data-driven methodology. Our approach integrates thousands of diverse genome-scale datasets by simultaneously using both tissue-specific and functional contexts. This technique effectively leverages signals detected by distinct technologies from experiments spanning both tissues and disease states. The tissue networks predict lineage-specific response of genes to perturbation, reveal changing functional roles of genes depending on tissue context, and illuminate meaningful disease-disease associations. We show that genes with nominally significant p-values in genome-wide association studies (GWAS) can be used in conjunction with tissue-specific networks to identify biologically important disease-gene associations, a procedure we term NetWAS. NetWAS identifies disease-associated genes more accurately than GWAS alone or an approach using a non-tissue-specific functional network. Our webserver, GIANT, (http://giant.princeton.edu) provides an interface to human tissue networks with multi-gene query capability, network visualization, analysis tools, and downloadable networks. GIANT also enables NetWAS reprioritization of users' GWAS results.
............................................................................................................................
SB P32: A modeling framework for generation of positional and temporal simulations of transcriptional regulation
David Knox1, Robin Dowell2
1University of Colorado Anschutz Medical Campus, 2University of Colorado
Abstract: We present a modeling framework aimed at capturing both the positional and temporal behavior of transcriptional regulatory proteins. There is growing evidence that transcriptional regulation is the complex behavior that emerges not solely from the individual components, but rather from their collective behavior, including competition and cooperation. Our framework describes individual regulatory components using generic action oriented descriptions of their biochemical interactions with a DNA sequence. All the possible actions are based on the current state of factors bound to the DNA. We developed a rule builder to automatically generate the complete set of biochemical interaction rules for any given DNA sequence. Off-the-shelf stochastic simulation engines can model the behavior of a system of rules and the resulting changes in the configuration of bound factors can be visualized. We compared our model to experimental data at well-studied loci in yeast, confirming that our model captures both the positional and temporal behavior of transcriptional regulation.
Method: Our goal was to integrate inherently dynamic aspects of transcriptional regulation, such as transcriptional interference, with the intuitive position based models. To this end, we constructed a modeling framework that leverages the power of Petri nets to describe the actions of various regulators and the extent or span of their influence. By treating the DNA as an ordered set of entities (nucleotides or groups of nucleotides) rather than a single molecular entity, we can generate models that grow linearly with the length of the DNA sequence being modeled. At the core of our framework is our stochastic rule builder, an application that can take in an arbitrary sequence and construct the complete set of coherent biochemical rules. Off-the-shelf stochastic simulation engines, such as Dizzy, can then simulate these rule sets. The simulations produce tremendous amounts of positional and temporal data, which can be converted into simple visualizations depicting the state of the DNA at each time step.
Results: We have developed a framework to create biologically realistic models of the mechanisms of transcriptional regulation. Based on this framework, we can model not only the steady-state behavior of transcription factor binding and nucleosome formation, but also the dynamics of components such as the transcriptional machinery. Our framework scales linearly, making it possible to simulate very large segments of DNA. The simulations record the state of the complex system of interactions at each time step. We have interpreted this state information to visualize the binding configuration of components along the DNA.
............................................................................................................................
SB P33: Mutation analysis pipeline for E. coli
Erin Boggess1, Liam Royce1, Yingxi Chen1, Laura Jarboe1, Julie Dickerson1
1Iowa State University
Next-generation sequencing is increasingly accessible and affordable, resulting in vast quantities of genomic data. Metabolic evolution benefits from next-gen sequencing because of the ability to sequence and compare entire genomes. In microbial engineering, metabolic evolution is an essential method for developing organisms with a desired phenotype. In an evolution experiment, variants with advantageous phenotypes emerge under strong selective pressure and displace the parent strain in a population. Improved fitness is attributed to acquired mutations that are identified by comparing the genomes of evolved and parent strains. The technique generates a strain with a desired phenotype, but requires further research to ascertain how mutations relate to the phenotype.
After genome sequencing, a bottleneck occurs in annotating mutations and interpreting their effects. Traditionally, mutations are manually annotated and those within coding regions are investigated for relation to fitness. While drastic mutations such as loss of function will be revealed, this ignores extragenic changes that can affect regulation. Furthermore, manual annotation and exploration is tedious and significantly increases time to a secondary round of metabolic evolution and other experimentation. The massive amount of sequence variation data generated in adaptive evolution experiments necessitates computational tools that assess mutation significance.
Here we provide a pipeline to facilitate and expedite mutation analysis in E. coli with the underlying ambition of mapping genomic changes to phenotype. Our pipeline leverages public genome databases, gene regulatory networks, metabolic pathways, and computational tools such as structure prediction software.
The pipeline begins by annotating and classifying a mutation, then assigning a priority given its predicted effect to structure and function. We use the E. coli regulatory network to discover other genes regulated by the mutated element. This expanded gene list is examined to identify associated enzymatic reactions in the E. coli metabolic network. When studying a set of mutations, expanded gene and reaction lists are collected and examined for overrepresented pathways and GO terms. Knowledge of altered reactions, affected pathways, and gene function commonalities is key to mapping mutations to phenotype.
When applied to E. coli experimental data from a metabolic evolution study for enhanced octanoic acid tolerance, we identified mutations in extragenic regions that would traditionally be overlooked. In addition, regulatory links extended our understanding of mutation effects, and prioritization assisted in focusing the follow-up research efforts.
The proposed methods benefit the research community by broadening the study of mutations and mechanisms of adaptation. Additionally, automating portions of comparative genomic analysis reduces the lifecycle of metabolic evolution studies. From an initial list of mutations and genomic positions, we provide researchers with a prediction of how that mutation affects the element in which it occurs, cellular implications, and prioritization so that efforts may be focused on the most relevant mutations.
............................................................................................................................
SB P34: Network-based model of oncogenic collaboration for prediction of drug sensitivity
Ted Laderas1
1Oregon Health & Science University
Tumorigenesis is a multi-step process, involving the acquisition of multiple oncogenic mutations that transform cells, resulting in systemic dysregulation that enables tumor proliferation. High throughput “omics” techniques allow rapid identification of these mutations with the goal of identifying treatments that target them. However, the multiplicity of oncogenes required for transformation (oncogenic collaboration) makes mapping treatments difficult. To make this problem tractable, we have defined oncogenic collaboration as mutations in genes that interact with an oncogene that may contribute to its dysregulation, a new genomic feature we term “surrogate mutations.” By mapping the mutations to a protein/protein interaction network, we can determine significance of the observed distribution using permutation-based methods. We identified significant surrogate mutations in oncogenes such as BRCA1 and ESR1 that are frequently observed across breast cancer cell lines. In addition, using Random Forest Classifiers, we show that these significant surrogate mutations predict drug sensitivity in breast cancer cell lines with a mean error rate comparable to the current standard; e.g., the PAM50 expression data (30.1% vs 29.1%). Our model has potential for integrating patient-unique mutations in predicting drug sensitivity, suggesting a potential new direction in precision medicine as well as a new model for drug development. Additionally, we show the prevalence of significant surrogate mutations in breast cancer patients within the Cancer Genome Atlas, suggesting that surrogate mutations may be a useful genomic feature in personalized medicine.
............................................................................................................................
SB P35: Emergence in signal transduction networks: identification of complex mechanisms of information transfer to understand and control cell phenotypes in health and disease
Mark Ciaccio1, Neda Bagheri1
1Northwestern University
Many drug candidates fail in clinical trials due to an incomplete understanding of how small-molecule perturbations affect signal transduction at the systems level. Small molecules can bind multiple proteins, exerting non-intuitive emergent effects on cell phenotype due to nonlinear signaling properties such as feedback and redundancy. We created a computationally-efficient algorithm, DIONESUS (Dynamic Inference Of NEtwork Structure Using Singular Values), based on partial least squares regression (PLSR) to accurately reconstruct the signaling network architecture from the phosphoproteomic signatures of 60 phosphosites at four time points following 30 diverse perturbations. This dynamic dataset was collected using the microwestern array, a high-confidence and high-throughput method for assaying protein abundance and modification.
DIONESUS enabled us to explore several questions that are central to computational modeling of cell signaling: How much predictive power is gained by (i) accounting for temporal dependence and information flow in network structure, (ii) characterizing non-linear interactions among nodes, and (iii) inferring non-additive relationships between parent and daughter nodes, such as AND-, OR-, and XOR-gates? Understanding the essential properties of signaling networks beyond linear combinations of variables provides insight into how biological networks process information to create controlled and robust responses from noisy stimuli. We integrated our new methodology with phosphoproteomic data to hypothesize new mechanisms of information transfer and propose viable drug targets that take into account the complex and emergent properties of signal transduction networks.
............................................................................................................................
SB P36: A first truly systems level mechanistic model – unravelling the gene regulation of Th2 differentiation
Mattias Köpsén1, William Lövfors1, Sören Bruhn2, Gunnar Cedersund1, Mikael Benson1, Mika Gustafsson1
1Linköping University, 2Karolinska Institute
Recent and ongoing revolutions in measurement technologies imply completely new possibilities for genome research; today, time-resolved, quantitative, and systems-level data are available. Nevertheless, without a corresponding revolution in methods for data analysis, these new data tend to drown researchers and doctors, rather than provide clear and useful insights. Such new methods are developed within the field of systems biology. Systems biology has two main approaches: mechanistically detailed and well-determined simulation models for small subsystems, and more approximative statistical models for the entire genome. However, there are few, if any, methods that combine the strengths of these two approaches. Herein, we present LASSIM, a new simulation-based approach, which can be applied to systems of the size of the entire genome. The superior performance of LASSIM is demonstrated in three examples: i) an example with simulated data shows that unlike traditional large-scale methods, LASSIM correctly identifies the true behavior between measured data-points, ii) LASSIM outperforms the winner of a previous DREAM challenge, the most competitive benchmarking approach available, iii) based on new data from TH2 differentiation, LASSIM identifies a first mechanistic model for the entire genome. The key predictions of this model are typically enriched for DNA bindings, which suggests that most predicted interactions are direct. Moreover, in silico knockdowns were experimentally validated. In summary, LASSIM opens the door to a new type of model-based data analysis: models that combine the strengths of reliable mechanistic models with truly systems-level data.
............................................................................................................................
SB P37: Utilization of Whole Genome Analysis Approaches for Personalized Therapy Decision Making in Patients with Advanced Malignancies
Martin Jones1, Yaoqing Shen1, Erin Pleasance1, Katayoon Kasaian1, Sreeja Leelakumari1, Yvonne Y Li1, Peter Eirew2, Richard Corbett1, Karen L Mungall1, Nina Thiessen1, Yussanne Ma1, Alexandra Fok1, Jacquie Schein1, Andrew J Mungall1, Yongjun Zhao1, Richard A Moore1, Stephen Yip3, Karen Gelmon4, Howard Lim4, Daniel Renouf4, Anna Tinker4, Sophie Sun4, Robyn Roscoe1, Steven JM Jones1, Janessa Laskin4, Marco A Marra1,5
1Canada's Michael Smith Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, BC, Canada; 2Department of Molecular Oncology, British Columbia Cancer Agency, Vancouver, BC, Canada; 3Centre for Translational and Applied Genomics, British Columbia Cancer Agency, Vancouver, BC, Canada; 4Department of Medical Oncology, British Columbia Cancer Agency, Vancouver, BC, Canada; 5Department of Medical Genetics, University of British Columbia, Vancouver BC, Canada
Genomic analysis is being widely investigated to support cancer treatment decision-making; here we detail our experience at the BC Cancer Agency. Our Personalized Oncogenomics (POG) project aims to determine the feasibility of a whole genome data driven approach to support these treatment decisions at a tertiary cancer care centre.
The POG project enrolls patients with metastatic cancers for which standard chemotherapy regimens fail or do not exist. For each patient, we performed whole genome (80-100X) and transcriptome sequencing (100-200M paired reads) of a fresh tumour biopsy sample and whole genome sequencing (40-50X) of peripheral blood. When available, the genome of an archival sample was also sequenced. All samples also undergo targeted deep sequencing using a “panel” to analyze selected known cancer mutations. Bioinformatics approaches were used to identify genes with somatic single nucleotide variants, small insertions and deletions, copy number variants, regions of loss of heterozygosity, structural variants, and expression changes to build an individual somatic molecular profile. This is followed by intensive pathway analysis and literature searches to identify the candidate biological processes that have been affected by mutation. Infectious agent presence and expression is also determined along with any integration sites.
Based on the integration of all these results, therapeutic options can be proposed. POG has consented 140 patients and sequenced and analyzed samples from 90 patients (including 6 pediatric) representing 33 different tumour types. The time between acquiring the biopsy sample and presenting a report for clinical oncologists was ~30-50 days. For ~70% of the patients the genome and transcriptome data were informative in guiding treatment. In addition the whole genome analysis approach was found to complement standard of care clinical tests aiding diagnosis.
Significant spatial and temporal tumour heterogeneity has often been noted as well as molecular response to treatment.
The POG project has now moved to a 2nd phase with ethical approval for 5,000 patients. Accrual has reached the rate of 5 patients per week with a ten fold increase expected over the coming 6-12 months.
Top of Page
REGULATORY GENOMICS PRESENTATIONS & ABSTRACTS
Presented on Thursday, November 13 and Friday, November 14
Updated Nov 11, 2014
--> Go directly to Friday, Nov 14
9:45 am – 10:05 am
RG T01
Identifying genetic and environmental determinants of gene expression
Roger Pique-Regi1, Christopher Harvey1, Gregory Moyerbrailean1, Omar Davis1, Donovan Watza1, Xiaoquan Wen2, Francesca Luca1
1Wayne State University, 2University of Michigan, Ann Arbor
The effect of genetic variants on a molecular pathway, and ultimately on the individual’s phenotype, is likely modulated by “environmental” factors. However, it is generally difficult to determine in which tissues and conditions genetic variants may have a functional impact. We denote the functional genetic variants that show cellular environment-specific effects as GxE expression quantitative trait loci (GxE-eQTLs). Achieving a better understanding of the mechanisms underlying GxE-eQTLs is a critical step in understanding the link between genotype and complex phenotypes.
To identify and characterize GxE-eQTLs we have established a new two-step and cost-effective experimental approach. In the first step, we identify global changes in gene expression using low-coverage sequencing of pools of highly multiplexed samples. In the second step, we select a subset of samples for deep sequencing and allele-specific analysis. For the first step, we generated 960 RNA-seq libraries in pools of 96 spanning 265 cellular environments across 5 cell-types (3 individuals), and 53 different treatments (including hormones, dietary components, environmental contaminants and metal ions). Relevant GO categories were enriched in the observed global gene expression changes (e.g., immune response for Dexamethasone, ion homeostasis for Zinc). We then analyzed allele specific expression (ASE) using a novel method (QuASAR) that allows for joint genotyping and allele specific analysis on RNA-seq data. Across 56 cellular environments we discovered 7738 instances of ASE (FDR<10%), corresponding to 6234 unique ASE genes. Using a Bayesian model across treatments within cell types, we observed that generally >95% ASE signals are shared and their effect sizes are highly concordant (posterior correlation coefficient 0.9). This is highly consistent with previous analysis of condition-specific eQTLs. Nevertheless, out of 112,564 tests we still estimate 2318 loci with a Bayes posterior probability supporting GxE interaction (1273 sites treatment-specific and 1045 sites control-specific, GxE-eQTLs). Genes that are differentially expressed also show a higher enrichment for condition-specific ASE. Our results constitute a first comprehensive catalog of GxE-eQTLs and we anticipate that it will contribute to the discovery and understanding of GxE interactions underlying complex traits.
...............................................................................................................................
Thursday, November 13
10:05 am – 10:25 am
RG T02
A pooling-based approach to mapping genetic variants associated with DNA methylation
Irene Kaplow1, Sarah Mah2, Julia MacIsaac2, Michael Kobor2, Hunter Fraser1
1Stanford University, 2University of British Columbia
DNA methylation is an epigenetic modification that plays a key role in gene regulation. Previous studies have investigated its genetic basis by mapping genetic variants that are associated with DNA methylation at specific sites, but these have been limited to microarrays that cover less than 2% of the genome and cannot account for allele-specific methylation (ASM). Other studies have performed whole-genome bisulfite sequencing on a few individuals, but these lack statistical power to identify variants associated with methylation. We present a novel approach in which bisulfite-treated DNA from many individuals is sequenced together in a single pool, resulting in a truly genome-wide map of DNA methylation. Compared to methods that do not account for ASM, our approach increases statistical power to detect associations while sharply reducing cost, effort, and experimental variability. As a proof of concept, we generated deep sequencing data from the pooled DNA of 60 human cell lines and identified over 2000 genetic variants associated with DNA methylation. We found that these variants are enriched in tissue-specific transcription factor binding sites and can also be associated with chromatin accessibility and gene expression. In sum, our approach allows genome-wide mapping of genetic variants associated with DNA methylation in any species, without the need for individual-level genotype or methylation data.
...............................................................................................................................
Thursday, November 13
10:25 am – 10:45 am
RG T03 • FULL LENGTH MANUSCRIPT
Are all genetic variants in DNase I sensitivity regions functional?
Gregory A. Moyerbrailean1, Chris T. Harvey1, Cynthia A. Kalita1, Xiaoquan Wen1, Francesca Luca1, Roger Pique-Regi1
1Wayne State University
A detailed mechanistic understanding of the direct functional consequences of DNA variation on gene regulatory mechanism is critical for a complete understanding of complex trait genetics and evolution. Here, we present a novel approach that integrates sequence information and DNase I footprinting data to predict the impact of a sequence change on transcription factor binding. Applying this approach to 653 DNase-seq samples, we identified 3,831,862 regulatory variants predicted to affect active regulatory elements for a panel of 1,372 transcription factor motifs. Using QuASAR, we validated the non-coding variants predicted to be functional by examining allele-specific binding (ASB). Combining the predictive model and the ASB signal, we identified 3,217 binding variants within footprints that are significantly imbalanced (20% FDR). Even though most variants in DNase I hypersensitive regions may not be functional, we estimate that 56% of our annotated functional variants show actual evidence of ASB. To assess the effect these variants may have on complex phenotypes, we examined their association with complex traits using GWAS and observed that ASB-SNPs are enriched 1.22-fold for complex traits variants. Furthermore, we show that integrating footprint annotations into GWAS meta-study results improves identification of likely causal SNPs and provides a putative mechanism by which the phenotype is affected.
...............................................................................................................................
Thursday, November 13
11:10 am – 11:30 am
RG T04
Viral and retrotransposon sequences have shaped the preferred contexts for APOBEC-mediated mutagenesis
Jeffrey Chen1, Thomas MacCarthy1
1Stony Brook University
The AID/APOBEC gene family of cytidine deaminases consists of mutagenic enzymes that have evolved roles in innate immunity such as virus restriction and suppression of transposable elements, particularly in mammals. The ancestral APOBEC gene, Activation Induced Deaminase (AID) arose early in vertebrate evolution and plays a key adaptive immunity role (somatic hypermutation of the Immunoglobulin genes) in all jawed vertebrates. Biochemical and in vivo profiling of many APOBECs shows they cause C to T transitions and have evolved a variety of local DNA sequence context preferences. APOBEC3F, for example, has a preference for mutations at TTC sites whereas APOBEC3G has a preference for CCC. We assess the impact of each motif on a set of potential target genes to investigate how individual preferences have been shaped. By specifically examining the impact of replacement mutations we demonstrate that the known APOBEC preferences maximally impact retrotransposons while minimally impacting essential host genes. Furthermore, permutation analysis of several mammalian virus genomes shows these have evolved to avoid the impact of these mutations. Our results also suggest that APOBEC preferences impose restrictions on codon and amino acid usage in their target genes by, for example, heavily disfavoring amino acid pairs that must encode the TTC motif favored by APOBEC3F.
...............................................................................................................................
Thursday, November 13
11:30 am – 11:50 am
RG T05
Quantitative modeling of transcription factor binding specificities using DNA shape
Tianyin Zhou1, Ning Shen2, Lin Yang1, Namiko Abe3, John Horton2, Richard Mann3, Harmen Bussemaker3, Raluca Gordan2, Remo Rohs1
1University of Southern California, 2Duke University, 3Columbia University
Our current knowledge of genome function is the result of sequence-based data in the form of one-dimensional strings of letters. However, DNA-binding proteins recognize the double helix as a three-dimensional object. Therefore, an understanding of transcription factor (TF) binding specificity must ultimately include DNA shape. The sequence-structure relationship in DNA is highly degenerate, and different nucleotide sequences can give rise to the same structure, while single nucleotide sequence variants sometimes change DNA shape over a region of several base pairs. To explore these effects on a genomic scale, we developed a method for the high-throughput DNA shape features. We used these structural features to augment nucleotide sequence in binding specificity models derived from statistical machine learning approaches such as support vector regression (SVR) and regularized multiple linear regression (MLR). Using these approaches, we learned in vitro DNA binding specificity models from protein binding microarray (PBM), genomic-context PBM, and HT-SELEX/SELEX-seq data. Based on data for many TFs from diverse protein families, we demonstrated that shape-augmented models are generally more efficient than existing sequence models in terms of accuracy, number of features, and computation time. Our models provide information on the importance of specific DNA sequence and shape features and thus reveal TF family-specific readout mechanisms and better explain why a given TF binds in vivo to a specific genomic target site.
...............................................................................................................................
Thursday, November 13
11:50 am – 12:10 pm
RG T06
Genome-wide map of regulatory interactions in the human genome
Nastaran Heidari1, Douglas Phanstiel1, Michael Snyder1
1Stanford University
Increasing evidence suggests that interactions between regulatory genomic elements play an important role in regulating gene expression. We generated a genome-wide interaction map of regulatory elements in human cells (ENCODE tier 1 cells, K562, GM12878) using Chromatin Interaction Analysis by Paired-End Tag sequencing (ChIA-PET) experiments targeting six broadly distributed factors. Bound regions covered 80% of DNase I hypersensitive sites including 99.7% of TSS and 98% of enhancers. Correlating this map with ChIP-seq and RNA-seq data sets revealed cohesin, CTCF, and ZNF143 as key components of three-dimensional (3D) chromatin structure and revealed how distal chromatin state affects gene transcription. Comparison of interactions between cell types revealed that enhancer-promoter interactions were highly cell-type specific. Construction and comparison of distal and proximal regulatory networks revealed stark differences in structure and biological function. Proximal binding events are enriched at genes with housekeeping functions while distal binding events interact with genes involved in dynamic biological processes including response to stimulus. This study reveals new mechanistic and functional insights into regulatory region organization in the nucleus.
...............................................................................................................................
Thursday, November 13
12:10 pm – 12:30 pm
RG T07
Deconvolution of massively- parallel reporter assays tiling 15,000 human regulatory regions reveal activating and repressive regulatory sites at nucleotide-level resolution
Jason Ernst1, Tarjei Mikkelsen2, Manolis Kellis3
1University of California, Los Angeles, 2Broad Institute, 3Massachusetts Institute of Technology
Massively parallel reporter assays have enabled genome-scale validation experiments towards gaining a systems-level view of gene regulation. A series of studies have demonstrated their use for testing thousands of predicted enhancers, dissecting regulatory motifs within them, and testing synthetically designed sequences. However, even with tens of thousands of sequences tested in a single assay, it has been impractical to dissect large numbers of regions at nucleotide level resolution, without an a priori knowledge of predicted regulatory motifs, limiting their large scale use to validation, but not discovery.
Here, we overcome this limitation, and present a new Bayesian tiling deconvolution approach, which combines experimental tiling of regulatory regions using 31 sequences of length 145 bp at 5 bp intervals covering 295 bp in total with computational deconvolution of the resulting signal to infer a nucleotide-level view of regulatory activity across thousands of regulatory regions. By exploiting the multiple overlapping sequences in a probabilistic framework, our method is also robust to noisy or missing measurements, and enables high-resolution inferences with a very small number of tested sequences per target region. This enables the de novo discovery of individual binding sites, and inference of their activating or repressive action in a single experiment across thousands of candidate regions. In contrast, activating and repressive sites are generally not distinguishable in current DNase hypersensitivity footprinting assays, as they both show footprints.
We apply this method to more than 15,000 regions in the human genome, in two ENCODE cell types, selected based on the presence of DNase hypersensitivity and chromatin marks covering a diverse range of regulatory regions, including enhancers, promoters, and insulator regions. Our method resulted in a regulatory activity score for more than 4.5 million nucleotides, which we used to predict bases of activation and repression. These nucleotides showed strong enrichments for motifs associated with activation or repression in the cell type.
Our method enables an unbiased, de novo, and high-resolution view of regulatory bases, which complements current motif scanning and DNase hypersensitivity footprinting approaches, and provides the first nucleotide-level view of activating and repressive sites across a sizeable fraction of the regulatory human genome.
...............................................................................................................................
Thursday, November 13
1:55 pm – 2:15 pm
RG T08 • FULL LENGTH MANUSCRIPT
cDREM: Inferring dynamic combinatorial gene regulation
Aaron Wise1, Ziv Bar-Joseph1
1Carnegie Mellon University
Motivation: Genes are often combinatorially regulated by multiple transcription factors (TFs). Such combinatorial regulation plays an important role in development and facilitates the ability of cells to respond to different stresses. While a number of approaches have utilized sequence and ChIP-based datasets to study combinational regulation, these have often ignored the combinational logic and the dynamics associated with such regulation.
Results: Here we present cDREM, a new method for reconstructing dynamic models of combinatorial regulation. cDREM integrates time series gene expression data with (static) protein interaction data. The method is based on a hidden Markov model and utilizes the sparse group Lasso to identify small subsets of combinatorially active TFs, their time of activation, and the logical function they implement. We tested cDREM on yeast and human data sets. Using yeast we show that the predicted combinatorial sets agree with other high-throughput genomic datasets and improve upon prior methods developed to infer combinatorial regulation. By applying cDREM to study human response to flu we were able to identify several combinatorial TF sets, some of which were known to regulate immune response while others represent novel combinations of important TFs.
...............................................................................................................................
Thursday, November 13
2:15 pm – 2:35 pm
RG T09
ATAC-seq is predictive of chromatin state
Chuan-Sheng Foo1, Sarah Denny1, Jason Buenrostro1, William Greenleaf1, Anshul Kundaje1
1Stanford University
Distinct combinations of chromatin modifications (chromatin states) have been found to be associated with different types of active and repressed functional elements in the human genome such as promoters, enhancers, and transcribed elements. Previously, multivariate hidden Markov models (e.g. ChromHMM and Segway) have been used to learn combinatorial chromatin states and automatically annotate genomes. However, such methods typically require multiple high-quality chromatin mark datasets as input, thus limiting their applicability in practice. Chromatin ChIP-seq experiments are time-consuming and costly to perform, and more importantly, require large amounts of input material to obtain reliable signal. We (Greenleaf lab) recently developed an assay, ATAC-seq, that accurately profiles genome-wide chromatin accessibility, DNA binding protein footprints, and nucleosome positioning from low amounts of input material based on direct in vitro transposition of sequencing adaptors into native chromatin. We previously showed that loci with different chromatin states (learned from histone modification ChIP-seq datasets) showed distinct distributions of ATAC-seq insert sizes in aggregate.
In this work, we further this connection between chromatin architecture and chromatin states by showing that chromatin architecture is in fact predictive of chromatin state at individual loci. More concretely, we show that a machine learning model trained on various features derived solely from ATAC-seq data is able to accurately predict different classes of regulatory elements in active and repressed chromatin states in cell lines and primary cells. The success of our method suggests that different classes of regulatory elements are associated with distinct open chromatin and nucleosome positioning signatures. We explore the feasibility of cross-cell-line chromatin state prediction and determine the minimum sequencing depth required for good predictive performance by subsampling reads. In conclusion, when applied to ATAC-seq data, our method enables high quality genome-wide chromatin state annotations from low quantities of input material using a single assay, potentially enabling the in vivo dissection of chromatin states from (rare) sorted cell populations in primary tissue.
...............................................................................................................................
Thursday, November 13
2:35 pm – 2:55 pm
RG T10
Integrative analysis of haplotype-resolved epigenomes across human tissues
Inkyung Jung1, Danny Leung1, Nisha Rajagopal1, Bing Ren1
1Ludwig Institute of Cancer Research
Allelic differences between the two sets of chromosomes can affect the propensity of inheritance in humans; however, the extent of such differences in the human genome has yet to be fully explored. Here, for the first time, we delineate allelic chromatin modifications and transcriptomes amongst a broad set of human tissues, enabled by a chromosome-spanning haplotype reconstruction strategy. The resulting masses of haplotype-resolved epigenomic maps are the first of its kind and reveal extensive allelic biases in the transcription of human genes, which appear to be primarily driven by genetic variations. Furthermore, allelic resolution of chromatin states allows us to discover cis-regulatory relationships between genes and their control sequences. These maps also uncover intriguing characteristics of cis-regulatory elements and tissue-restricted activities of repetitive elements. The rich datasets described here will enhance our understanding of the mechanisms controlling tissue-specific gene expression programs.
...............................................................................................................................
Thursday, November 13
2:55 pm – 3:15 pm
RG T11
Mechanistic basis of causal non-coding FTO obesity variant
Melina Claussnitzer1, Simon Dankel2, Gerald Quon3, Han Kim4, Hans Hauner5, Manolis Kellis3
1Harvard Medical School, 2University of Bergen, 3Massachusetts Institute of Technology, 4University of Toronto, 5Technical University Munich
Genome-wide association (GWA) studies revealed thousands of non-coding complex trait and disease genetic associations, whose mechanistic underpinnings remain elusive. Here, we leverage the Roadmap and ENCODE epigenomic maps across diverse human tissues and cell types to gain new insights into the regulatory underpinnings of the strongest genetic association with risk to polygenic obesity — i.e., the FTO obesity-associated locus — as a model for deciphering non-coding complex trait genetic associations. We find that the obesity-associated region harbors a >10kb super-enhancer candidate active in the adipose lineage, suggesting a role of adipose in FTO locus activity. We narrow down the causal region from 47kb and 82 variants ultimately to a single-nucleotide causal variant and identify its long-distant downstream target genes located up to a million nucleotides away in adipocytes. Using a comparative motif module analysis approach, we show that the causal variant overlaps a cluster of cross-species conserved regulatory motif instances for predicted master regulators enriched in obesity-associated variants across the genome, and that the risk allele disrupts the binding of the predicted transcriptional repressor, which is highly expressed in adipose cells. We demonstrate regulator-dependent repression of both the enhancer and its target genes in adipocytes conditional on the risk allele. We further confirm that the identified single-nucleotide change results in cellular phenotypes consistent with obesity, including decreased mitochondrial energy dissipation and increased triglyceride accumulation, based on repressor-dependent and allele-conditional differences in primary human adipocytes. Lastly, we evaluate the obesity role of the identified causal variant at the organismal level in transgenic mice, by generating an adipose-specific inhibition of target gene activity, opposite to their tissue-specific risk allele-dependent de-repression by the variant, and find dramatic differences in body weight, fat accumulation, and mitochondrial energy expenditure. Overall, our results suggest that an intronically located non-coding variant is the causal variant underlying the FTO association with obesity, by disrupting regulator-mediated repression of the identified long-distant target genes, and resulting in an adipose-specific shift from mitochondrial energy production to energy storage. We propose that the FTO locus controls energy dissipation in the form of heat, in a cell-autonomous way, opening up the potential to new therapeutics that directly target mitochondrial activity in adipocytes by exploiting the cell-regulatory circuitry of repressor, variant, and target that we have unraveled in this study. Overall, we here introduce a general model for the elucidation of non-coding variants associated with complex traits and disease, including: (1) establish the relevant tissue and cell type; (2) establish the target genes; (3) establish the causal variant; (4) recognize the upstream regulator; (5) establish the cellular phenotypic consequences; and (6) establish the organismal phenotypic consequences.
...............................................................................................................................
Thursday, November 13
3:40 pm – 4:00 pm
RG T12
Systematic detection of spatio-temporal patterns of epigenetic changes
Petko Fiziev1, Constantinos Chronis1, Kathrin Plath1, Jason Ernst1
1University of California, Los Angeles
Histone modifications associate with important regulatory regions such as promoters and enhancers that control the expression of genes. Time-course genome-wide maps of these epigenetic marks have become available in a growing number of biological settings, including somatic cell reprogramming and differentiation processes, circadian rhythms, embryogenesis, and lymphocyte development. However, our understanding of the underlying cellular processes remains limited because the current bioinformatics tools often fail to utilize fully the temporal aspects of this data. Here, we present a novel computational method for systematic detection of major classes of spatio-temporal patterns of epigenetic changes. The method takes as input data a series of chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) experiments for a single histone mark that are performed at consecutive time points during a given biological process. The method uses a probabilistic mixture model that explicitly models the spatio-temporal nature of the data to identify regions for which the mark either expands or contracts significantly with time or holds steady. We present applications of our method on data from somatic cell reprogramming in mouse and from cardiac differentiation that help in understanding the regulatory dynamics of these processes.
...............................................................................................................................
Thursday, November 13
4:00 – 4:20 pm
RG T13
MyoD induces active and poised chromatin structures during transdifferentiation
Dinesh Manandhar1, Lingyun Song1, Ami Kabadi1, Charles Gersbach1, Raluca Gordan1, Greg Crawford1
1Duke University
Overexpression of transcription factor (TF) MyoD has been shown to transdifferentiate cells from non-myogenic lineages into cells with muscle-like expression and functional characteristics. However, expression studies show that the transdifferentiated cells have only some myogenic genes upregulated. Chromatin level reprogramming is also incomplete. In this work, we investigate the reasons behind incomplete MyoD-induced transdifferentiation of fibroblasts, including potential MyoD cofactors, DNA methylation, and posttranslational histone modifications. We analyzed high-throughput chromatin accessibility (DNase-seq) data, in vivo MyoD binding (ChIP-seq) data, and global gene expression (RNA-seq) data on primary skin fibroblast cells transduced with inducible MyoD, and compared against the data obtained from starting fibroblast cells and target myoblasts and myotubes. Our study of local chromatin changes genome-wide suggests that the chromatin state of transdifferentiated fibroblasts is intermediary between fibroblast and muscle chromatin states. Importantly, we observed a continuum of chromatin reprogramming in the MyoD-induced fibroblasts, indicating that complete reprogramming is achieved in only a small fraction of the genome. We also see evidence that during MyoD-induced transdifferentiation, chromatin closes more easily than it opens up. Using random forest and support vector machine classifiers, we show that various genetic and epigenetic features dictate the efficiency of chromatin level reprogramming. For instance, fibroblast DNase hypersensitive sites (DHSs) with higher GC content tend to stay open more than DHSs with low GC content. Our analysis of TF motifs and histone modification data suggests that the presence of certain TFs or histone modification marks at or around a genomic site can dictate the efficiency of chromatin reprogramming. Analysis of gene expression data shows that reprogramming of genes correlates well with reprogrammed chromatin state. Nonetheless, enriched levels of “poised” or “memory” state chromatin are also observed around such genes. This indicates that MyoD is capable of inducing both active and poised chromatin structures that are similar to primary muscle lineages, and that other additional factors – such as Uhrf1, a chromatin remodeler under-expressed in transdifferentiated cells – can potentially help improve the reprogramming efficiency. Interestingly, we also found that although MyoD binding in non-DHSs opens up the chromatin at many genomic loci, a big fraction of MyoD-bound sites remain closed. Most of these closed sites lack MyoD-specific binding sites, which suggests that during transdifferentiation MyoD can also bind non-specifically or mediated by protein cofactors.
...............................................................................................................................
Thursday, November 13
4:20 pm – 4:40 pm
RG T14: Epigenomics of the Mammalian Brain
Chongyuan Luo1
1Joseph Ecker Laboratory, The Salk Institute for Biological Studies
The mammalian brain consist of numerous neuronal and non-neuronal cell types that are functionally indispensable. The abundances of distinct cell types can differ by orders of magnitude. The absence of representation for sparse, but nevertheless functionally critical, cell types in whole tissue genomic analyses presents a challenge. We generated an extensive epigenomics dataset from nuclei of specific neuronal types purified by INTACT (isolation of nuclei tagged in specific cell types) approach. We identified abundant epigenomic and regulatory differences in nearly 200,000 discrete regions across pyramidal neurons, parvalbumin (PV)-expressing and vasoactive intestinal peptide (VIP)-expressing interneurons in adult mouse. Non-CG DNA methylation accounts for nearly half of all DNA methylation in adult mouse neurons and inversely correlates with cell-type specific gene expressions. Significant interactions between putative transcription factor binding and epigenomic features suggest an interplay between sequence specific and non-specific mechanisms for maintaining neuronal type specification. Our results stress the importance of cell-type specific methods for studying epigenomes and identification of gene regulatory mechanisms in complex tissues such as the mammalian brain.
...............................................................................................................................
Top of Page
9:45 am – 10:05 am
RG15
Cell type-specific regulatory networks reveal common pathways of disease variants
Gerald Quon1, Melina Claussnitzer1, Michal Grzadkowski1, Manolis Kellis1
1Massachusetts Institute of Technology
Genome-wide association studies (GWAS) have identified thousands of single nucleotide variants associated with diverse human traits, but understanding their combined action in complex systems remains an open challenge. With more than 80% of lead GWAS SNPs located in non-coding regions of the genome rich in regulatory elements, functionally characterizing these variants necessitates knowledge of (1) the locations of cell type specific enhancers; (2) the identity of the target genes of those enhancers; and (3) the interactions between these target genes to identify disrupted pathways and subnetworks. However, existing gene-centric networks may not be suitable for network analysis because of uncertainty over the target genes of non-coding GWAS loci.
Using enhancer and promoter maps for 127 cell types predicted by the Roadmap Epigenomics Consortium, we have constructed directed cell type specific networks, where nodes represent four types of elements: transcriptional regulators, non-coding regulatory elements (enhancers, promoters), SNPs, and target genes. Edges lead from transcription factors to regulatory elements, and regulatory elements to genes, while SNPs are connected to tagged regulatory elements. To leverage these networks for GWAS analysis, we developed an efficient probabilistic model to simultaneously (1) identify the target regulatory element of each individual non-coding GWAS locus, from among the set of all tagged elements; (2) identify TF regulators whose binding sites are characteristic of GWAS target regulatory elements and distinguish them from other active regulatory elements in the cell type; and (3) identify other regulatory elements likely to harbor additional weak effect GWAS variants.
We applied our networks and model to a diverse range of GWAS traits, including metabolic (diabetes), lipid, and brain disorders. We find that our predicted regulators of complex traits recapitulate many known regulators from the literature, and we have experimentally validated several novel regulators in type 2 diabetes. Our cross validation experiments holding out subsets of GWAS loci during model training demonstrate that our networks have predictive power for identifying GWAS variants. We also found that based on network structure alone, a subset of sub-genome wide significant GWAS loci exhibit exceptionally strong evidence for involvement in trait variation, and their predicted target genes also are implicated in the complex trait. Finally, we show target genes of predicted target regulatory elements of GWAS loci yield sensible phenotypes in mouse when mutated. These results taken together suggest that these regulatory element-centric networks, combined with our novel probabilistic model, can help yield insight into the important genomic players of complex traits and disease.
...............................................................................................................................
Friday, November 14
10:05 am – 10:25 am
RG T16
Assessing the impact of non-silent somatic mutations on protein activity
Mariano Alvarez1, Federico Giorgi1, Yao Shen1, Andrea Califano1
1Columbia University
Large-scale sequencing of cancer genomes often reveals thousands of non-silent (amino acid-changing) somatic mutations (NSSM) in proteins. However, not all cancer mutations affect the molecular function of the mutated protein. Current computational approaches to differentiate functional from non-functional variants are based on predicting physiochemical effects of the substitutions, taking into account the protein surface placement in interaction sites, secondary and tertiary structure features, and evolutionary conservation of the affected protein domains. There is no approach, however, to estimate the effect of somatic mutations on protein activity in an unbiased and genome-wide fashion.
In this work, we measured the association between non-silent somatic mutations and the activity of the mutated protein, as estimated by the VIPER algorithm, for 3,912 tumor samples, representing 14 different tumor types, for which matched gene expression and exome or genome profile data is available from TCGA. For this, we assembled tissue type-specific regulatory models and inferred the activity of each transcription factor and signaling protein in each sample using the VIPER algorithm. We then tested whether samples harboring NSSMs in specific proteins were enriched among those ranked by the VIPER-inferred protein activity.
To illustrate the potential of our approach, we focused the analysis on 147 genes listed by the Catalogue Of Somatic Mutations In Cancer (COSMIC), which were mutated in at least 2 samples within the same tumor type. We find significant association of the mutations with VIPER-inferred protein activity for 75 of the 147 proteins (p < 0.05). Interestingly, NSSM were also significantly associated with differential expression for 85 of the evaluated genes. To isolate the independent contribution of protein activity (i.e., the purely post-translational effect), we eliminated any transcriptional component from VIPER-inferred activities by removing the transcriptional variance component from the analysis. Remarkably, we found significant association between NSSM and transcriptional-independent VIPER-activity for 71 of the 147 tested proteins, showing that the effect of mutations is largely independent of transcriptional changes in the corresponding genes and that VIPER analysis can effectively capture these post-transcriptional, NSSM-dependent effects.
Finally, we increased the resolution of the analysis by testing whether different NSSMs within the same host gene (e.g., G12V vs. G12D KRAS mutations) may differentially affect protein activity. In total, we analyzed 648 NSSMs affecting 49 genes across 12 tumor types. Careful analysis of these results showed that VIPER-detected changes in protein activity are both mutation-specific and tumor type-specific.
To our knowledge, this work constitutes the first genome-wide and unbiased approach to catalogue the functional relevance of coding somatic mutations in cancer, with profound implications in the discovery of driver variants at the single patient level, and clear application in precision medicine.
...............................................................................................................................
Friday, November 14
10:25 am – 10:45 am
RG T17 • FULL LENGTH MANUSCRIPT
A validated gene regulatory network and GWAS to identify early transcription factors in T-cell associated diseases
Mika Gustafsson1, Danuta Gawel1, Sandra Hellberg1, Aelita Konstantinell1, Daniel Eklund1, Jan Ernerudh1, Antonio Lentini1, Robert Liljenström1, Johan Mellergård1, Hui Wang2, Colm E. Nestor1, Huan Zhang1, Mikael Benson1
1Linköpings UniveristetUniversitet, 2MD Anderson Cancer Center
The identification of early regulators of disease is important for understanding disease mechanisms, as well as finding candidates for early diagnosis and treatment. Such regulators are difficult to identify because patients generally present when they are symptomatic, after early disease processes. Here, we present an analytical strategy to systematically identify early regulators by combining gene regulatory networks (GRNs) with GWAS. We hypothesized that early regulators of T-cell associated diseases could be found by defining upstream transcription factors (TFs) in T-cell differentiation. Time-series expression profiling identified upstream TFs of T-cell differentiation into Th1/Th2 subsets enriched for disease associated SNPs identified by GWAS. We constructed a Th1/Th2 GRN based on integration of expression, DNA methylation profiling, and sequence-based predictions data using LASSO algorithm. The GRN was validated by ChIP-seq and siRNA knockdowns. GATA3, MAF, and MYB were prioritized based on GWAS and the number of GRN predicted targets. The disease relevance was supported by differential expression of the TFs and their targets in profiling data from six T-cell associated diseases. We tested if the three TFs or their splice variants changed early in disease by exon profiling of two relapsing diseases, namely multiple sclerosis and seasonal allergic rhinitis. This showed differential expression of splice variants of the TFs during relapse-free asymptomatic stages. Potential targets of the splice variants were validated based on expression profiling and siRNA knockdowns. Those targets changed during symptomatic stages. Our results show that combining construction of GRNs with GWAS can be used to infer early regulators of disease.
...............................................................................................................................
Friday, November 14
11:10 am – 11:30 am
RG T18
Enhancer RNAs reveal widespread chromatin reorganization in prostate cancer cell lines
Ville Kytölä1, Annika Kovakka1
1University of Tampere
Chromatin conformation determines the gene regulatory programs and enables the diversity of cell types. Characterization of chromatin state across different cell lines has been a central focus of major projects such as ENCODE. These studies have revealed a number of insights into cellular programs in cell differentiation and disease related dysregulation. However, the degree of chromatin variation between individuals is less studied and the diversity of chromatin organization in cancer is not known.
In order to gain insight into diversity of chromatin organization in prostate cancer we characterized 11 prostate and prostate cancer cell lines under different culture conditions using Global Run-On sequencing (GRO-seq). This assay allowed us to identify the active enhancer areas from each cell line through detection of nascent transcription of enhancer RNA (eRNA) molecules. To this end, we developed a new computational algorithm to identify eRNA signals in a genome-wide manner by utilizing the unique bi-directional pattern of nascent transcription. Identified eRNA sites show high consistency with areas of open chromatin from DNase I sequencing (DNase-seq) data as over 80% of the sites are covered by open chromatin signals in LNCaP cells.
We present a comparison of eRNA signals across prostate cancer cell lines. Our analysis reveals extensive variation in enhancer activity between prostate cancer models. On average, approximately 3000 active eRNA loci were identified from each cell line with the number of detected sites varying from 1300 to 8000. Based on the detection results, the cell lines clustered according to androgen receptor (AR) status. When cultured in the presence of androgens, the number of identified eRNA sites in LNCaP and VCaP cells doubled in comparison to cells cultured without androgens. Overall, we identified nearly 25,000 distinct loci of which only 33% were shared between more than two cell lines. We find a high number of loci for which eRNA activity correlates with the expression of nearby genes. Interestingly, from among these sites we were able to extract a subset of over a hundred extremely highly correlating ( > 0.9) connections, strongly indicating that these enhancer regions are contributing to the phenotypic diversity of prostate cancer. Taken together, these analyses highlight several new patterns of active enhancer regions that associate with specific prostate cancer subtypes. We are integrating eRNA activities with DNA methylation and transcriptome data from the same cell lines to uncover detailed regulatory programs in prostate cancer.
...............................................................................................................................
Friday, November 14
11:30 am – 11:50 am
RG T19
Identifying differential functions of cancer mutations using a structurally resolved protein interaction network
Hatice Billur Engin1, Matan Hofree1, Hannah Carter1
1University of California, San Diego
Here we present a method for discovering the distinct functional outcomes of different somatic missense mutations in a protein. This is the first attempt, to our knowledge, to explicitly account for diverse structural consequences of mutations for protein activity when extracting altered gene sets from tumor ‘omics data. Until now, efforts to mine large tumor ‘omics datasets have assumed that all damaging amino acid substitutions in a protein have the same consequences for protein activity, and that all of the protein’s interactions are equally impacted. However, disease-causing mutations are frequently observed at interface residues mediating protein interactions[1-4]. These residues are not essential for protein stability but are nontrivial for the binding energies of protein-protein interactions. As a result, mutations at interfaces may cause diverse phenotypes by changing the interaction profile of the mutated protein.
In this study, we generated a hybrid protein-protein interaction network with a subset of edges that include protein structural information for frequently mutated cancer genes. By mapping missense mutations reported by The Cancer Genome Atlas onto the 3D structures of the encoded proteins, we identified core and interface mutations. We used these designations to alter the network by removing 1 or more edges according to whether the mutation is more likely to destabilize specific interaction(s) or the entire protein. Then, using a diffusion-based approach on the altered network, we implicated distinct sets of interacting proteins associated with different mutated residues. The interacting proteins for each mutation were functionally annotated to highlight specific biological processes likely to be affected.
Although the number of 3D structures capturing protein-protein interactions is small, our analysis using the existing structures provides strong evidence supporting specific functional consequences of somatic missense mutations at distinct sites within the same protein. We performed an in-depth case study of HRAS, implicating distinct biological processes associated with mutations observed at residues G12, G13 and G61 that may have clinical relevance for patients. We also clustered cancer patients based on diffusion profiles incorporating residue-specific effects to find subgroups of patients that are similar at the level of disrupted biological processes. Our analysis suggests that accounting for mutation-specific perturbations to cancer pathways will be essential for personalized cancer therapy.
References:
1 David, A., Razali, R., Wass, M. N. & Sternberg, M. J. Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Human mutation 33, 359-363, doi:10.1002/humu.21656 (2012).
2 Wang, X. et al. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nature biotechnology 30, 159-164, doi:10.1038/nbt.2106 (2012).
3 Yates, C. M. & Sternberg, M. J. The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein-protein interactions. Journal of molecular biology 425, 3949-3963, doi:10.1016/j.jmb.2013.07.012 (2013).
4 Zhong, Q. et al. Edgetic perturbation models of human inherited disorders. Molecular systems biology 5, 321, doi:10.1038/msb.2009.80 (2009).
...............................................................................................................................
Friday, November 14
11:50 am – 12:10 pm
RG T20
Network-based Stratification of Tumor Profiles
Matan Hofree1, John P. Shen2, Hannah Carter3, Andrew Gross3, Trey Ideker1,2,3
1Department of Computer Science and Engineering, University of California San Diego, CA
2Department of Medicine, University of California San Diego, CA
3Department of Bioengineering, University of California San Diego, CA
Classification of cancer is predominantly organ based and fails to account for considerable heterogeneity of clinical outcomes such as survival or response to therapy. Somatic tumor genomes provide a rich new source of data for uncovering subtypes, but have proven difficult to compare, as tumors rarely share the same mutations. Recently we introduced network-based stratification (NBS), a method to integrate somatic tumor genomes with gene networks. This approach allows for stratification of cancer into subtypes by clustering together patients with perturbations in similar network regions. We demonstrate NBS in multiple cancer cohorts from The Cancer Genome Atlas. In each case, NBS identifies subtypes that are predictive of clinical outcomes such as patient survival, response to therapy or histology. We show that subtypes may be reproduced in an independent cohort and are predictive of chemo-resistance in cell-lines. Finally, we show how we can integrate somatic mutations, gene fusion events and copy-number changes to discover subtypes of thyroid cancer and identify network regions characteristic of each type.
...............................................................................................................................
Friday, November 14
1:00 pm – 1:20 pm
RG T21 • FULL LENGTH MANUSCRIPT
Multi-species network inference improves gene regulatory network reconstruction for early embryonic development in Drosophila
Anagha Joshi1, Yvonne Beck1, Tom Michoel1
1The Roslin Institute, The University of Edinburgh
Gene regulatory network inference uses genome-wide transcriptome measurements in response to genetic, environmental, or dynamic perturbations to predict causal regulatory influences between genes. We hypothesized that evolution also acts as a suitable network perturbation and that integration of data from multiple closely related species can lead to improved reconstruction of gene regulatory networks. To test this hypothesis, we predicted networks from temporal gene expression data for 3,610 genes measured during early embryonic development in six Drosophila species and compared predicted networks to gold standard networks of ChIP-chip and ChIP-seq interactions for developmental transcription factors in five species. We found that (i) the performance of single-species networks was independent of the species where the gold standard was measured; (ii) differences between predicted networks reflected the known phylogeny and differences in biology between the species; (iii) an integrative consensus network which minimized the total number of edge gains and losses with respect to all single-species networks performed better than any individual network. Our results show that in an evolutionarily conserved system, integration of data from comparable experiments in multiple species improves the inference of gene regulatory networks. They provide a basis for future studies using the numerous multi-species gene expression datasets for other biological processes available in the literature.
...............................................................................................................................
Friday, November 14
1:20 pm – 1:40 pm
RG T22
Comparison of D. melanogaster and C. elegans developmental stages, tissues, and cells by modENCODE RNA-Seq data
Jingyi (Jessica) Li1, Haiyan Huang2, Peter J. Bickel2, Steven Brenner2
1University of California, Los Angeles, 2University of California, Berkeley
We report a statistical study to discover transcriptome similarity of developmental stages from D. melanogaster and C. elegans using modENCODE RNA-seq data. We focus on "stage-associated genes" that capture specific transcriptional activities in each stage and use them to map pairwise stages within and between the two species by a hypergeometric test. Within each species, temporally adjacent stages exhibit high transcriptome similarity, as expected. Additionally, fly female adults and worm adults are mapped with fly and worm embryos, respectively, due to maternal gene expression. Between fly and worm, an unexpected strong collinearity is observed in the time course from early embryos to late larvae. Moreover, a second parallel pattern is found between fly prepupae through adults and worm late embryos through adults, consistent with the second large wave of cell proliferation and differentiation in the fly life cycle. The results indicate a partially duplicated developmental program in fly. Our results constitute the first comprehensive comparison between D. melanogaster and C. elegans developmental time courses and provide new insights into similarities in their development. We use an analogous approach to compare tissues and cells from fly and worm. Findings include strong transcriptome similarity of fly cell lines, clustering of fly adult tissues by origin regardless of sex and age, and clustering of worm tissues and dissected cells by developmental stage. Gene ontology analysis supports our results and gives a detailed functional annotation of different stages, tissues, and cells. Finally, we show that standard correlation analyses could not effectively detect the mappings found by our method.
...............................................................................................................................
Friday, November 14
1:40 pm – 2:00 pm
RG T23
Context-specific regulation by miR-155 through ApA-dependent and independent mechanisms
Gabriel Loeb1,2, Yuheng Lu1, Jing-Ping Hsin12, Christina Leslie1, Alexander Rudensky1,2
1Memorial Sloan Kettering Cancer Center, 2Howard Hughes Medical Institute
MicroRNAs (miRNAs) are critical post-transcriptional regulators of gene expression that repress expression of target mRNAs by mediating the interaction between RISC and cognate sites in 3’UTRs. Recent studies have begun to investigate whether miRNAs, like transcription factors, regulate their targets in a cell-type and context dependent manner. For example, alternative polyadenylation (ApA) produces cell-type specific changes in 3’UTR isoform expression, and relative “shortening” or “lengthening” of 3’UTRs can lead to loss or gain of miRNA binding sites. Previous studies, however, have reached conflicting conclusions about the impact of ApA on the cell-type specificity of miRNAs. Moreover, it is plausible that there are ApA-independent mechanisms of miRNA context specificity, such as the tissue-specific expression of particular RNA-binding proteins.
miR-155 is an important regulator in the immune system and is up-regulated upon activation of multiple immune cell types, including macrophages, dendritic cells, and T and B lymphocytes. To examine the cell-type specificity of miR-155 in physiologically relevant cellular contexts, we performed PolyA-seq and RNA-seq in these four activated immune cell populations from WT and miR-155 KO mice. Importantly, PolyA-seq enables the analysis of miR-155 regulation at the level of 3’UTR isoforms as well as detection of ApA between cell types. Quantitative expression analysis revealed that a large fraction of miR-155 targets are significantly differentially regulated between cell types at both the gene and 3’UTR isoform levels. Overall, miR-155 targets are strongly enriched for genes with multiple 3’UTR isoforms. Among the multi-isoform target genes, there is a significant overlap between genes differentially regulated by miR-155 and genes exhibiting ApA between cell types. In-depth analysis suggested that ApA-dependent miRNA specificity may be a combination of two mechanisms: (1) lengthening or shortening of 3’UTRs can result in gain or loss of miRNA target sites; and (2) the regulatory activity of the same miRNA target site may depend on its relative position within 3’UTR isoforms. Meanwhile, we have also found that in many cases the same target 3’UTR isoform can be differentially regulated between cell types, suggesting ApA-independent mechanisms of miRNA specificity.
...............................................................................................................................
Friday, November 14
2:00 pm – 2:20 pm
RG T24 • FULL LENGTH MANUSCRIPT
Systematic study of synthetic transcript features in S. cerevisiae exposes gene-expression determinants
Tuval Ben-Yehezkel1, Shimshi Atar2, Tzipy Marx1, Rafael Cohen1, Alon Diament2, Alexandra Dana2, Anna Feldman2, Ehud Shapiro1, Tamir Tuller2
1Weizmann Institute of Science, 2Tel Aviv University
A major challenge in functional genomics is understanding how different parts of the transcript affect aspects of its expression. Heterologous gene expression can potentially contribute to this research topic, but has rarely been studied systematically, specifically in eukaryotes.
Here, we use a synthetic biology approach to study the distinct and causal effect of different parts of the transcript in the eukaryote S. cerevisiae. We generated three distinct reporter libraries of the viral HRSVgp04 gene for studying the effect of three distinct regions in the transcript: (1) the 5'UTR, (2) the first 40 codons, and (3) codons 42-81of the ORF. Each of the three libraries contained variants with multiple, rationally designed synonymous mutations, totaling 383 distinct variants tested individually for gene expression.
Our results show that while synonymous mutations in each of the three regions can have a dramatic effect on protein abundance, those closer to the 5’ end of the ORF are the most effective modulators of protein abundance. Additionally, while weaker local mRNA folding at the beginning of the ORF (codons 1–8) increases protein abundance, it decreases protein abundance when present in downstream codons, reinforcing previous evolutionary studies demonstrating the selection of folding strength in different parts of the ORF. Finally, we show that the mean relative codon decoding time, based on ribosomal densities in endogenous genes, significantly correlates with our measured protein abundance (correlation up to r = 0.6175; p=0.0013). While this report provides an improved understanding of transcript evolution and gene expression regulation, it also suggests relatively simple rules for engineering synthetic gene expression in a eukaryote.
Top of Page | Go directly to Friday, Nov 14






















