
ISMB 2020 - Special Sessions
- SST01: Systems Immunology: Computational approaches for understanding human immune system and immune-related diseases
- SST02: Bioinformatics outside the lab: How to mobilize online citizen scientists to accelerate research
- SST03: Bioinformatics of Corals
- SST04: SCANGEN: Single-cell cancer genomics
- SST05: HuBMAP: Integrating genomics, imaging and mass-spectrometry to construct single-cell human tissue maps
- SST06: Interpreting the Lipidome – Approaches to Embrace the Complexity
- SST07: The Brain Initiative Cell Census Network
SST01: Systems Immunology: Computational approaches for understanding human immune system and immune-related diseases (abstract submission details at http://sysimm.lji.org/ )
Monday, July 13 10:40 am - 6:00 pm
Organizer(s):
Ferhat Ay, La Jolla Institute for Allergy and Immunology, United States
Duygu Ucar, The Jackson Laboratory for Genomic Medicine, University of Connecticut Health Center, United States
Presentation Overview:
Dysfunctions in the immune system are important contributing factors, if not the driving factors, for a wide range of human diseases. Advances in next generation sequencing techniques are providing us opportunities to closely monitor human immune cells in health and disease, and uncover genomic and epigenomic signatures that can be linked to pathologies. However, the analyses and integration of multi-faceted genomic data are not trivial and require sophisticated computational methods. These methods are developed for data processing and analysis (e.g., imaging, cell sorting, epitope identification, single-cell sequencing), for linking genetic and epigenetic variation to disease susceptibility (GWAS, eQTL, chromatin QTL) and for integrating epigenome, transcriptome and cell abundance measurements from multiple distinct immune cells in the context of immune related diseases such as autoimmune diseases and cancer. As the amount of data collected from individuals to profile their immune systems is increasing, we are encountering a bottleneck to integrate cross-platform data and mine these to test biological hypotheses. This meeting aims to bring together computational scientists who develop methods to effectively analyze single-cell level and bulk measurements from human immune cells in diverse contexts including aging, autoimmune diseases and cancer as well as in the broader context of understanding genetic and epigenetic regulation of gene expression.
SST02: Bioinformatics outside the lab: How to mobilize online citizen scientists to accelerate research
Monday, July 13, 2:00 pm - 6:00 pm
Organizer(s):
Jérôme Waldispühl, McGill University, Canada
This meeting aims to offer a comprehensive overview of the progress and achievements of citizen science applications to biology over the past 10 years. Each talk will discuss a different challenge and describe a solution made possible by the application of crowdsourcing to a specific biological research question.
- The origin of scientific games in molecular biology. This talk will discuss the genesis of the Foldit project and early results that pioneered the field of scientific discovery games for molecular biology. It will provide an overview of the foundation of this field of research.
- Building a knowledge base for biologists. The curation and organization of biological knowledge is an essential resource for researchers, but this task cannot always be fully automated. We will discuss the opportunities offered by crowdsourcing approaches to building and maintaining databases.
- Collaborative design of new molecules. We will discuss how citizen scientists collaborate to discover new scientific principles, and describe algorithmic frameworks allowing to harness the work of crowds.
- When gamers meet experimentalists. Scientific games allow users to process raw experimental data and submit hypothesis to scientists that can be validated in labs. This theme will explore opportunities arising with the creation of new bridges between citizens and scientists.
- Building communities of citizen scientists. The success of citizen science initiatives relies on the capacity to build and train large communities of participants. This talk aims to explore strategies to promote engagement and their impact on the performances.
- How to turn scientific tasks into casual games. In this talk, we will show how complex computational tasks can be embedded in casual games in order to make them accessible to broader audiences.
- Extreme citizen science: Scaling-up citizen science with video games and AI. The integration of citizen science tasks in real video games can help to reach very quickly large crowds of participants not naturally exposed to science. Then, the application of AI techniques can also help to exploit the large volume of data collected.
Tuesday July 14, 10:40 am - 12:40 pm
Organizers
Lenore Cowen, Department of Computer Science, Tufts University, United States
Judith Klein-Seetharaman, Department of Chemistry, Colorado School of Mines, United States
Hollie Putnam, Department of Biological Sciences, University of Rhode Island, United States
Presentation Overview
Corals are important natural resources that are key to the oceans vast biodiversity and provide economic, cultural, and scientific benefits. As a result of anthropogenic activities, locally and globally, coral reefs are declining rapidly. The environmental sensitivity and symbiotic biological complexity of corals makes understanding the genomic variability that influences vulnerability and resilience of local coral reef systems very challenging. Corals are made up of thousands of different organisms, including the animal host and single celled dinoflagellate algae, bacteria, viruses, and fungi that coexist as a holobiont. Thus, corals are more like cities than individual animals, as they provide factories, housing, restaurants, nurseries, and more for an entire ecosystem, both at the micro and macro levels.
A large amount of genomic, transcriptomic and other omics data from different species of reef building corals, the uni-cellular dinoflagellates, plus coral microbiome data (where corals have possibly the most complex microbiome yet discovered, consisting of over 20,000 different species), is becoming increasingly available for corals. This is a terrific opportunity for bioinformatics researchers and computational biologists to contribute to a timely, compelling and urgent investigation of critical factors that influence reef health and resilience.
We have invited some of the premier experts who are working on bioinformatics of coral reefs to participate in our invited sessions. We will introduce this exciting topic to the ISMB community, with the goal of energizing collaborations and approaches to address the compelling problems in this captivating and complex system. This convergence of data and critical need to address this declining ecosystem provides a timely and impactful topic for ISMB 2020.
SST04: SCANGEN: Single-cell cancer genomics (abstract submission details at http://www.scangen.org/)
Tuesday July 14, 2:00 pm - 6:00 pm
Organizer(s):
Kieran R Campbell, Lunenfeld-Tanenbaum Research Institute, University of Toronto, Canada
Presentation Overview:
The past decade has resulted in technological advances that have given us the unprecedented ability to measure RNA, DNA, and epigenetic modifications at the single-cell level, combined with widespread acquisition of imaging data. This has enabled routine measurement of genomic, morphologic and histopathologic alterations across tens of thousands of cells, discovering new cell types, developmental lineages, and cell-specific mutational patterns. This new data has prompted an explosion in statistical and computational methods development (http://www.scrna-tools.org/) with over 540 tools created in the past five years alone.
However, the majority of methods developed remain focused on either technical aspects (such as normalization and differential expression) or on applications in developmental biology such as lineage inference and cell clustering, with relatively little attention applied to the huge potential of single-cell data to unveil the complex biology behind cancer initiation and progression. As one of the first workshops of its kind, this special session will bring together researchers developing computational and statistical methods for single-cell cancer biology. It will focus around (though not be limited to) four core topics:
- Modeling cancer evolution
As tumors evolve they accumulate both point mutations and large structural rearrangements. The “life-histories” of these tumors are informative of the mutational processes that allow the cancer cells to evade the body’s checkpoints and can be predictive of future evolution and response to therapy. Methods covered under this topic could address: phylogenetic inference from single-cell data; inference of evolutionary processes from single-cell data; identifying singlecell cancer signatures; inference of fitness from single-cell analysis of population dynamics. - Integrative analyses of multi-modal and imaging data
A vast array of measurements can be made at single-cell resolution, including RNA and DNA-sequencing and epigenetic status such as methylation and chromatin accessibility. Methods covered in this topic will include: modelling of joint measurement assays (such as G&T-seq); relating and interpreting measurements from different technologies. - Scalable inference at the single-cell level
A typical single-cell RNA or DNA-seq dataset now contains around 100x more cells than it did less than a decade ago. As a result, there is a pressing need for computational and statistical methods that scale to “big data” sizes, particularly since fast computation allows iterative analyses by investigators, aiding biological interpretation. Methods covered in this topic will include: scalable statistical inference for single-cell data using methods such as stochastic optimization; computational tools for dealing with large single-cell datasets. - Interactions and perturbations at the single-cell level
This broad topic concerns methods to understand how cancer cells react to both their environment and external perturbation. Methods could address: how cells interact with their microenvironment; how cells respond to and resist chemotherapeutic interventions; how transcriptional programming and clonal selection are affected by genomic perturbations such as CRISPR.
SST05: HuBMAP: Integrating genomics, imaging and mass-spectrometry to construct single-cell human tissue maps
Tuesday July 14, 2:00 pm - 6:00 pm
Organizers
Ziv Bar-Joseph, Carnegie Mellon University, United States
Nils Gehlenborg, Harvard Medical School, United States
Ajay Pillai, National Human Genome Research Institute, United States
Presentation Overview
Transformative technologies are enabling the construction of three-dimensional maps of tissues with unprecedented spatial and molecular resolution. Over the next six years, the NIH Common Fund Human Biomolecular Atlas Program (HuBMAP) intends to develop a widely accessible framework for comprehensively mapping the human body at single-cell resolution by supporting technology development, data acquisition, computational analysis and detailed spatial mapping. The HuBMAP project exists within a larger ecosystem of other single cell atlas projects and this session will cover both.
In this special session we will focus on the computational goals, needs and current state of the HuBMAP project. After introducing the types of single cell resolution data HuBMAP proposes to generate a series of speakers will present specific solutions to the following challenges: (1) infrastructure for accessing the data; (2) tools and pipelines for analysis and integration of data; (3) visualizing and querying single cell data at scale; (4) creating a reference common coordinate framework (CCF) to spatially represent single-cell data mapped to specific organs and systems will be addressed.
The intended audience for this special session includes computational researchers that focus on the analysis and integration of large scale biological data. We would both discuss methods, and unmet needs for the analysis of single cell sequence data (scRNA-Seq, scATAC-Seq, etc.), spatial transcriptomics (microscopy) and proteomics (imaging mass spectrometry) data. While the initial pipelines and tools developed for HuBMAP are being implemented by consortium members, we expect that both, additional analysis and additional tools would come from other members of the community who would have access to the data via the web interface and a set of dedicated APIs.
Thursday, July 16, 3:20 pm - 4:40 pm
Organizer(s):
Bobbie-Jo Webb-Robertson, Pacific Northwest National Laboratory, United States
Jason McDermott, Pacific Northwest National Laboratory, United States
Geremy Clair, Pacific Northwest National Laboratory, United States
Presentation Overview
Due to the central role that lipids play in energy metabolism, structure and signaling, current research programs ranging from biomedical to ecological applications have shown an increase in the analysis of lipids [1]. Lipidomics specifically is the large-scale study of the structure and function of all lipids in a sample, termed the lipidome, as well as the interaction of lipids with other biomolecules, including proteins, metabolite and other lipids. Research employing lipidomics is rapidly increasing. As seen in Figure 1, the number of publications referring to lipidomics or the lipidome in their title in the National Library of Medicine PubMed database has increased by over 2-fold in just the last 5 years. This is because recent advancements in mass spectrometry (MS) technology together with bioinformatic developments allow more researchers to perform studies with lipids. However, because the field of lipidomics is younger than the other omics fields and the structure, diversity and complexity of the lipidome differs from than these predecessor omics there is considerable opportunity to advance the field through computation. 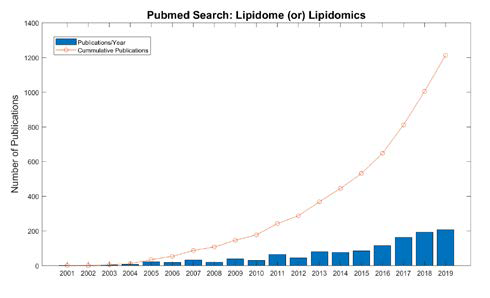
The International Lipid Classification, and National Nomenclature Committee referred to as LIPID MAPS (http://www.lipidmaps.org) [2] began by categorizing lipids into eight categories based on their chemical and biochemical properties. These classes are further divided by more specific structural and chemical properties. As of January 2018 there are over 40,000 lipid structures in this database and it is expected to continue to grow [3]. This complexity in the lipidome is often under-described by averaging lipids up to the class-based descriptors, losing valuable information about biological function and interactions with other lipids, proteins or metabolites.
Although lipidomics is considered a sub-field of metabolomics it is much earlier in methods development than for metabolite identification and evaluation [4] and the computational needs are still a mystery to many working in bioinformatics and computational biology. While methods and tools have improved over the past few years, computational developments are at the center of the gap in deriving insights from this data. The complexity of lipidomics data means that global-based analyses only identify hundreds of lipids versus the thousands present. In addition, inferring the interaction of the lipidome with the genome and proteome is key to mechanistic understanding.
ISMB participants are interested in cutting edge research and areas of great need for new developments, thus this session will be of interest to a large collection of attendees. To see lipidomics reach its full potential improved strategies for identification and quantification, as well as pathway and integration approaches are needed.
Thursday, July 16, 3:20 pm - 4:40 pm
Organizers
Jesse Gillis, Cold Spring Harbor Laboratory, United States
Presentation Overview
The mammalian brain is a complex organ relying on a circuitry involving millions to billions of cells. Identifying neuronal cell types and how these cell types interact is an essential step toward understanding the organization of the brain. The Brain Initiative Cell Census Network (BICCN) aims to define a taxonomy of robust neuronal cell types that can be characterized through multiple modalities, e.g. transcriptional similarity, spatial organization, morphology and connectivity. In the past three years, the BICCN has generated several large reference molecular datasets intended to serve as a broad reference for understanding and analyzing the mammalian brain. As a complex system with an unusual number of modalities to define cell identity, the brain serves as a potential Rosetta stone to clarify many pressing questions regarding the properties and functions of cell types. In this session, we will describe essential properties of the available BICCN data and walk through best practices in its use. As a reference data set on par with the largest previous NIH consortium efforts, the BICCN promises to be an essential resource for future research by data focused scientists in biology.
The BICCN is a very large NIH initiative. While significant outreach is planned (and has been undertaken) to the neuroscience community, with multiple successful symposia by many of the same speakers planned for this special session, there has been little planned outreach to the bioinformatics community. Indeed, with the exception of the Gillis lab, none of the planned speakers have previously attended ISMB. While the analyses from the BICCN are likely to be of broad interest (and currently being submitted as a series of consortium papers, largely available on biorxiv by the time of ISMB2020), its true main contribution is the data generated, available for use by others. For this resource to be fully exploited, the evaluation and characterization by the BICCN needs to be shared with a broader community of data researchers. This special session is intended to make this a reality.
